一种三酮类化合物的合成方法与流程
1.本发明属于有机合成领域,具体一种三酮类化合物(二甲酰亚胺类化合物)的合成方法。
背景技术:
2.截至目前,制备该类二甲酰亚胺类化合物通常使用的方法就是通过苯甲酰异氰酸酯和烯醇的反应。arbuzov,b.a.等人(ba arbuzov,zobova n n,dautova l a.cheminform abstract:reaction of acyl(aroyl)isocyanates with dimedone[j].chemischer informationsdienst,1982,13(20):115
‑
116)将苯甲酰异氰酸酯与3
‑
羟基
‑
5,5
‑
二甲基环己酮在60~70℃反应生成三酮类化合物,收率51%,并同时生成25%的副产物。
[0003][0004]
上述已知方法的缺点是,反应易于产生副产,或者反应时间长,转化率低,导致收率低;或者使用了不安全的试剂,三废排放量大,进而导致环境污染。
技术实现要素:
[0005]
本发明目的在于提供一种合成工艺简单、转化率高、收率高的由烯醇酯反应重排生成三酮类除草剂(二甲酰亚胺类化合物)的合成方法。
[0006]
为实现上述目的,本发明采用技术方案为:
[0007]
一种三酮类除草剂的合成方法,由烯醇酯于溶剂中,在氰酸盐或硫氰酸盐的存在下,发生反应并进行重排,获得目标物三酮类化合物。
[0008]
进一步的说,式ii的所示的烯醇酯于溶剂中,在含氰酸盐或硫氰酸盐的溶剂的存在下,使式ii的所示的烯醇酯和氰酸盐或硫氰酸盐于溶剂中发生反应并重排,获得式i所示的三酮类化合物;其中,式i和式ii化合物结构式如下,
[0009][0010]
其中,
[0011]
r1、r2分别选自c1‑
c4烷基、c1‑
c4烷氧基,或者r1、r2与其相连的碳原子组成含碳或氧的五、六、七元环,如下结构所示:
[0012][0013]
y、y1、y2、y3、y4、y5、y6、y7、y8相同或不同的选自o、s或
[0014]
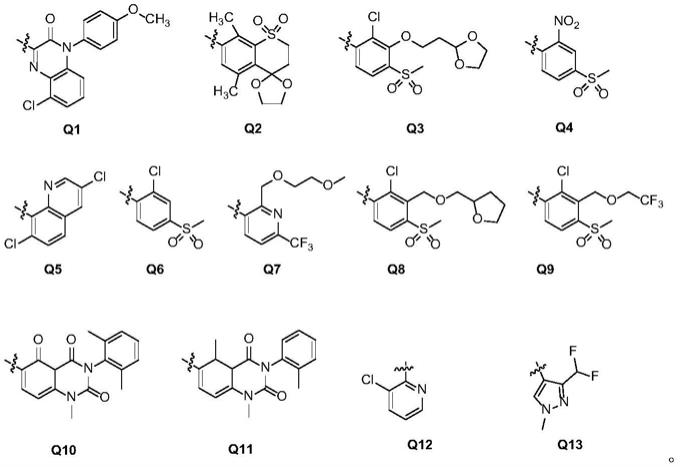
q选自如下所示基团之一:
[0015][0016]
所述氰酸盐或硫氰酸盐与式ii的烯醇酯的摩尔比值为1.0~2.0;优选1.1~1.5;
[0017]
所述溶剂为极性有机溶剂或非极性有机溶剂;
[0018]
进一步的说,所述溶解式ii的所示的烯醇酯、溶解氰酸盐或硫氰酸盐以及发生反应并重排时采用的溶剂可相同或不同的极性有机溶剂或非极性有机溶剂
[0019]
所述极性有机溶剂为乙腈、n,n
‑
二甲基甲酰胺、n,n
‑
二甲基乙酰胺、n
‑
甲基吡咯烷酮、二甲基亚砜、六甲基磷酰三胺(hmpa)、四氢呋喃、甲基异丁酮、乙酸乙酯、1,3
‑
二甲基
‑2‑
咪唑啉酮。
[0020]
所述非极性有机溶剂为:甲苯、二甲苯、氯苯、二氯甲烷、二氯乙烷、氯仿、四氯化碳。
[0021]
所述的氰酸盐或硫氰酸盐为氰酸钠(sodium cyanate)、氰酸钾、氰酸铵、氰酸钙、氰酸铯、氰酸镁、氰酸亚铁、氰酸铁、氰酸镍、氰酸铜、硫氰酸钠、硫氰酸钾或硫氰酸铵中的一种或几种。
[0022]
所述的重排反应在温度20~80℃下进行,反应时间为0.5~5小时;优选20~60℃下进行,反应时间为1~2.5小时。
[0023]
所述重排反应的转化速度和反应时间与温度有关,温度低,反应时间长,温度高,反应时间短,但采用本发明方法通常为1~2小时。
[0024]
所述的重排反应在非极性有机溶剂中进行时,可加入相转移催化剂以加速反应;其中,相转移催化剂加入量为烯醇酯质量的1~30%(质量分数)。
[0025]
所述相转移催化剂可选自本领域常规相转移催化剂,如peg
‑
200、peg
‑
400、peg
‑
600、18冠
‑
6、15冠
‑
5、环糊精、苄基三乙基氯化铵、四丁基溴化铵、四丁基氯化铵、四丁基硫酸氢铵、四甲基溴化铵、三丁基甲基氯化铵、三辛基甲基氯化铵、十二烷基三甲基氯化铵、十四烷基三甲基氯化铵、吡啶、三丁胺、甲基三苯基溴化磷、四丁基溴化磷、1,8
‑
二氮杂二环十一碳
‑7‑
烯(dbu)或三乙烯二胺中的一种或两种。
[0026]
上述制备过程中对于产物的含量以高效液相色谱采用外标法测定。
[0027]
另外,本发明所采用的烯醇酯原料(式ii的化合物)以及氰酸盐、硫氰酸盐和/或相转移催化剂均可以通过市售或是按照现有技术记载制备得到。
[0028]
本发明所具有的优点:
[0029]
本发明方法使用氰酸盐或硫氰酸盐的单一试剂在有机溶剂中在较短时间内完成烯醇酯到三酮类目标化合物的重排反应,进而避免反应体系中加入过多试剂,导致操作繁琐、产生副产,或者反应时间长,转化率低,导致收率低的问题;同时避免使用不安全的试剂,从而减少三废排放量,保护环境;使用便宜易得的氰酸盐或硫氰酸盐,进一步简化工艺,有效降低生产成本;本发明反应体系不易产生副产,转化率高,该方法易于工业化生产。
[0030]
本发明的部分化合物可以用表1中列出的具体化合物来说明,但并不限定本发明。
[0031]
表1
[0032]
[0033]
[0034]
具体实施方式
[0035]
以下具体实施例用来进一步说明本发明,但本发明绝非仅限于这些例子;并且下述实施例中所涉及的百分比均为质量百分比,如含量、纯度等。
[0036]
本发明所采用的原料(式ii的化合物)以及氰酸盐、硫氰酸盐和/或相转移催化剂均可以通过市售或是按照现有技术记载制备得到。
[0037]
实施例1
[0038][0039]
将氰酸钠2.34g(0.036mol)和50ml n,n
‑
二甲基甲酰胺加入反应瓶中,然后向其中滴加9.81g(0.03mol)烯醇酯ii
‑1‑
1的30ml n,n
‑
二甲基甲酰胺溶液,加毕,室温搅拌反应2小时,hplc监测反应完全,反应液倒入冰水中,用稀盐酸调节ph至1.5左右,固体过滤,水洗晾干,得到8.40g淡黄色固体i
‑1‑
1,含量97%(hplc归一)。收率
‑
75.6%。熔点:124
‑
126℃。
[0040]
实施例2
[0041][0042]
将氰酸钠2.15g(0.033mol)和50ml二甲基亚砜加入反应瓶中,然后向其中滴加10.20g(0.03mol)烯醇酯ii
‑1‑
6的30ml二甲基亚砜溶液,加毕,室温搅拌反应1小时,hplc监测反应完全,反应液倒入冰水中,用稀盐酸调节ph至1.5左右,固体过滤,水洗晾干,得到10.49g黄色固体i
‑1‑
6,含量98%(hplc归一)。收率91.5%。熔点:212
‑
214℃。
[0043]
实施例3
[0044][0045]
将氰酸钠10.73g(0.165mol)和300ml n,n
‑
二甲基甲酰胺加入反应瓶中,然后向其中滴加50.85g(0.15mol)烯醇酯ii
‑1‑
6的100ml n,n
‑
二甲基甲酰胺溶液,加毕,室温搅拌反应2小时,hplc监测反应完全,反应液倒入冰水中,用稀盐酸调节ph至1.5左右,固体过滤,水洗晾干,得到52.20g黄色固体i
‑1‑
6,含量99%(hplc归一)。收率91.1%。
[0046]
实施例4
[0047][0048]
将氰酸钠10.73g(0.165mol)和300ml n,n
‑
二甲基甲酰胺加入反应瓶中,然后向其中滴加56.77g(0.15mol)烯醇酯ii
‑2‑
10的100ml n,n
‑
二甲基甲酰胺溶液,加毕,室温搅拌反应1.5小时,hplc监测反应完全,反应液倒入冰水中,用稀盐酸调节ph至1.5左右,固体过滤,水洗晾干,得到55.89g白色固体i
‑2‑
10,含量99%(hplc归一),收率88.5%。熔点:181
‑
183℃。
[0049]
实施例5
[0050][0051]
将氰酸钠10.73g(0.165mol)和300ml n,n
‑
二甲基甲酰胺加入反应瓶中,然后向其中滴加66.0g(0.15mol)烯醇酯ii
‑1‑
11的100ml n,n
‑
二甲基甲酰胺溶液,加毕,室温搅拌反应1.5小时,hplc监测反应完全,反应液倒入冰水中,用稀盐酸调节ph至1.5左右,固体过滤,水洗晾干,得到63.46g黄色固体i
‑1‑
11,含量98%(hplc归一),收率87.6%。熔点:145
‑
147℃。
[0052]
实施例6
[0053][0054]
将氰酸钾14.58g(0.18mol)和200ml n
‑
甲基吡咯烷酮加入反应瓶中,然后向其中滴加37.73g(0.15mol)烯醇酯ii
‑3‑
2的80ml n
‑
甲基吡咯烷酮溶液,加毕,室温搅拌反应1小时,hplc监测反应完全,反应液倒入冰水中,用稀盐酸调节ph至1.5左右,固体过滤,水洗晾
干,得到37.11g黄色固体i
‑3‑
2,含量97%(hplc归一),收率84.02%。熔点:129
‑
131℃。
[0055]
另外,将反应式中通过改变原料的不同取代基,并依照上述制备过程的记载,还可以获得不同取代基所示的式i化合物,这也显示出了本发明方法应用的广泛性。
[0056]
本发明的其他化合物可以参照以上实施例制备。
[0057]
部分化合物的物性数据及核磁数据(1hnmr,600mhz,内标tms,ppm)如下:
[0058]
化合物i
‑1‑
1:黄色固体,熔点124
‑
126℃。δ(cdcl3)12.49(s,1h,nh),8.88
–
8.80(m,1h,phenyl
‑3‑
h),8.41
–
8.32(m,1h,phenyl
‑6‑
h),7.73
–
7.65(m,1h,phenyl
‑5‑
h),3.18
–
3.16(s,3h,so2ch3),2.50(s,3h,coch3),2.26(s,3h,ch3)。
[0059]
化合物i
‑1‑
6:黄色固体,熔点212
‑
214℃。δ(cdcl3)12.61(s,1h,nh),8.79(d,j=1.7hz,1h,phenyl
‑3‑
h),8.29(dd,j=7.9,1.7hz,1h,phenyl
‑6‑
h),7.63(d,j=7.9hz,1h,phenyl
‑5‑
h),3.17(s,3h,ch3),2.69(t,j=6.4hz,2h,cyclohexyl
‑
5,5
‑
2h),2.61
–
2.53(m,2h,cyclohexyl
‑
3,3
‑
2h),2.05
–
1.98(m,2h,cyclohexyl
‑
4,4
‑
2h)。
[0060]
化合物i
‑1‑
10:白色固体,熔点246
‑
248℃。δ(cdcl3)12.11(s,1h,nh),8.84(d,j=1.7hz,1h,phenyl
‑3‑
h),8.34(dd,j=7.9,1.7hz,1h,phenyl
‑6‑
h),8.04(dd,j=8.0,1.6hz,1h,phenyl
‑5‑
h),7.78(ddd,j=8.7,7.3,1.6hz,1h,phenyl
‑6‑
h),7.70(d,j=7.9hz,phenyl
‑5‑
h),7.44
–
7.39(m,2h,phenyl
‑
7,8
‑
2h),3.19(s,3h,ch3)。
[0061]
化合物i
‑1‑
11:黄色固体,熔点145
‑
147℃。δ(cdcl3)12.49(s,1h,nh),8.78(d,j=2.0hz,1h,phenyl
‑3‑
h),8.28(dd,j=8.0,1.9hz,1h,phenyl
‑6‑
h),7.63(t,j=8.3hz,1h,phenyl
‑5‑
h),3.96
–
3.79(m,2h,nch2),3.21
–
3.13(s,3h,so2ch3),2.74(t,2h,ch2),1.55(s,9h,3ch3)。
[0062]
化合物i
‑2‑
6:黄色固体,熔点139
‑
141℃。δ(cdcl3)12.73(s,1h,nh),8.01(d,j=1.9hz,1h,phenyl
‑3‑
h),7.91(dd,j=8.1,1.8hz,1h,phenyl
‑6‑
h),7.65(d,j=8.1hz,1h,phenyl
‑5‑
h),3.10
–
3.07(s,3h,ch3),2.73(t,j=6.6hz,2h,cyclohexyl
‑
5,5
‑
2h),2.61
–
2.47(m,2h,cyclohexyl
‑
3,3
‑
2h),2.03(dd,j=8.5,5.1hz,2h,cyclohexyl
‑
4,4
‑
2h)。
[0063]
化合物i
‑2‑
10:白色固体,熔点181
‑
183℃。δ(cdcl3)12.23(s,1h,nh),8.09(dd,j=8.0,1.6hz,1h,phenyl
‑5‑
h),8.06(d,j=1.7hz,1h,phenyl
‑3‑
h),7.96(dd,j=8.0,1.7hz,1h,phenyl
‑6‑
h),7.79(dd,j=8.7,7.3,1.6hz,1h,phenyl
‑6‑
h),7.74(d,j=8.0hz,1h,phenyl
‑5‑
h),7.46
–
7.42(m,1h,phenyl
‑7‑
h),7.41(d,j=8.5hz,1h,phenyl
‑8‑
h),3.12(s,3h,ch3)。
[0064]
化合物i
‑3‑
2:黄色固体,熔点129
‑
131℃。δ(cdcl3)12.74(s,1h,nh),8.52(dd,1h,pyridyl
‑6‑
h),7.89
–
7.85(m,1h,pyridyl
‑4‑
h),7.44
–
7.35(m,1h,pyridyl
‑5‑
h),2.74(t,j=6.4hz,2h,cyclohexyl
‑
5,5
‑
2h),2.62
–
2.53(m,2h,cyclohexyl
‑
3,3
‑
2h),2.10
–
2.00(m,2h,cyclohexyl
‑
4,4
‑
2h)。
[0065]
化合物i
‑4‑
3:白色固体,熔点235
‑
237℃。δ(cdcl3)12.39(s,1h,nh),8.13(dd,j=8.1,1.9hz,1h,phenyl
‑5‑
h),8.02(s,1h,pyrazolyl
‑5‑
h),7.85
–
7.76(m,1h,phenyl
‑6‑
h),7.51
–
7.44(m,1h,phenyl
‑7‑
h),7.45
–
7.40(m,1h,phenyl
‑8‑
h),7.04(t,1h,chf2),4.05(s,3h,ch3)。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1