一种封端苯并噁嗪树脂、包含其的双邻苯二甲腈基复合材料与复合固化树脂
1.本发明涉及苯并噁嗪领域,尤其涉及一种封端苯并噁嗪树脂、包含其的双邻苯二甲腈基复合材料与复合固化树脂。
背景技术:
2.随着我国大飞机、新能源、轨道交通等新兴行业的发展,热固性树脂基复合材料的应用领域不断拓展,其回收问题也越来越突出。国内外许多研究机构已纷纷提出了相关的课题,对其回收问题进行研究[化信.宁波材料所复合材料绿色回收研究获进展[j].化工新型材料,2013(2):168-168.]。
[0003]
对于热固性树脂的降解回收,国内外许多专家学者针进行了相关研究,主要方法包括物理法和化学法两种,其中物理法主要采用机械粉碎回收,化学法主要包括热解法和溶剂法[徐平来,李娟,李晓倩.热固性树脂基复合材料的回收方法研究进展[j].工程塑料应用,2013,41(1):100-104.]。山西煤化所侯相林团队利用配位不饱和的锌离子选择断裂环氧树脂的碳氮键,实现了碳纤维增强环氧树脂的高效降解及循环利用;利用弱配位的铝离子选择性断裂酯键,实现了玻璃纤维增强不饱和聚酯树脂的降解回收[deng t s,liu y,cui x j,et al.cleavage of c-n bonds in carbon fiber/epoxy resin composites.green chemistry,2015,17,2141-2145.]。t.iwaya等在二乙二醇单甲醚及苯乙醇的溶剂中,在k3po4的催化下,在190~350℃下反应1~8h,可降解不饱和聚酯,回收得到长玻璃纤维[iwaya t,et al.recycling of fiber reinforced plastics using depolymerization by solvothermal reaction with catalyst.j mater sci,2008,43(7):2452
–
2456.]。
[0004]
我国对热固性复合材料废弃物的处理主要采取填埋和焚烧,填埋的方法占用土地资源且造成土壤破坏。焚烧不会造成土地浪费,但由于燃烧中产生大量毒气,会造成二次污染,同时存在潜在的、未知的危险[化信.宁波材料所复合材料绿色回收研究获进展[j].化工新型材料,2013(2):168-168.]。
[0005]
因此,发展具有可分解的热固性树脂体系,是废弃的热固性树脂及其粘合剂、涂层材料、复合材料实现循环利用有效途径,也是热固性树脂领域发展的重要方向之一。
技术实现要素:
[0006]
苯并噁嗪树脂(bz)是以酚类、醛类、胺类化合物为原料,经缩合反应得到的含氮、氧六元杂环的化合物,是一类新型的热固性树脂,经过固化后得到的材料
‑‑
苯并噁嗪树脂除了具备传统酚醛树脂良好的耐热性和阻燃性以外,由于在开环固化过程中没有挥发性小分子放出,热固化收缩率接近为零,固化后的聚苯并噁嗪树脂孔隙率较低,内应力和裂纹几乎都不存在,有利于成品的加工成型及保持制品的尺寸,同时,苯并噁嗪树脂中几乎不存在游离的醛和酚,可作为机舱的阻燃材料,广泛应用于建筑、交通、航空、航天、电子、船舶、能
源等各个领域[郭军.国内外复合材料废弃物回收技术与发展现状[j].科技创新导报,2011(33):99-100.]。
[0007]
与此同时,苯并噁嗪仍存在一些缺点,如固化温度高,一般都要达到200℃;固化时间长;传统的苯并噁嗪聚合后得到的苯并噁嗪树脂较脆,机械性能不是很高;加工过程繁杂,大部分苯并噁嗪单体为固体,它们在加工过程中难以像液态热固性树脂预聚体那样方便地使用;预聚体分子量较低,很难加工成薄膜。另一方面,高分子量线性结构是一把双刃剑。随着材料性能的提高,制备和加工变得困难。预聚物在合成阶段容易形成凝胶,这使得反应时间控制和溶剂选择变得重要而复杂。另一方面,即使在非常高的温度下,大多数mcbp仍具有很高的粘度和不适合常规熔体加工的特性。迄今为止,由于溶液流延,衍生自mcbp的聚苯并噁嗪大多以薄膜形式出现。
[0008]
苯并噁嗪树脂固化后的交联网络结构不溶不熔,在回收和可降解方面极大的限制了它们的应用。如何能够把固化后的苯并噁嗪重新降解回收再利用是一个很现实的问题。在国家政策的指引下,应大力发展能耗小、回收效果好的热固性碳纤维复合材料废弃物工业化回收及再利用工艺,实现复合材料废弃物的资源化回收再利用,对建设资源节约型、环境友好型和谐社会,响应国内外保护环境、节能减排、可持续发展的号召具有重要意义[刘建叶,宋金梅,彭玉刚,等.热固性碳纤维复合材料废弃物回收及再利用现状[j].化工新型材料,2014(8):216-218.]。
[0009]
为了克服上述问题,本发明公开了一种封端苯并噁嗪树脂、包含其的双邻苯二甲腈基复合材料与复合固化树脂,所述具有主链结构的封端苯并噁嗪树脂为含有螺环缩醛结构的低分子量主链,该苯并噁嗪树脂及包含其的复合材料或固化物可以在酸性或高温下降解,对降解产物的分离、回收之后,可以实现材料的循环利用,从而完成本发明。
[0010]
本发明的目的之一是提供一种封端苯并噁嗪树脂,其为主链型聚合物,所述主链型聚合物的分子链中含有式(i-1)~式(i-3)所示重复单元中的至少一种:
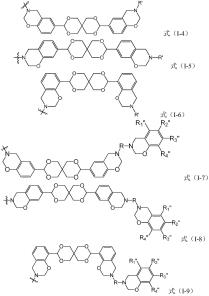
[0011][0012]
所述主链型聚合物的分子链端为式(i-4)~式(i-9)之一所示结构:
[0013][0014][0015]
在式(i-1)~式(i~9)中,每个r各自独立地选自脂肪基及其衍生物、脂环基及其衍生物、芳香基及其衍生物中的一种;和/或,在式(i-4)~式(i-6)中,r
′
选自脂肪基及其衍生物、脂环基及其衍生物、芳香基及其衍生物中的一种;和/或,在式(i-7)~式(i-9)中,r1″
~r4″
各自独立地选自氢、脂肪基及其衍生物、脂环基及其衍生物、芳香基及其衍生物中的一种。
[0016]
其中,在所述封端苯并噁嗪树脂中含有缩醛结构,可以赋予材料酸敏性,在酸性条件下实现降解,有利于材料的循环利用,具有良好的环境效益。进一步地,本发明采用的缩醛结构为螺环缩醛,其为闭合六元环结构,具有一定刚性,赋予苯并噁嗪树脂固化过程较好的耐热性,以及赋予其固化树脂耐热性和刚性。
[0017]
在一种优选的实施方式中,在式(i-1)~式(i~9)中,每个r各自独立地选自c2~c20的烷基及其衍生物、c6~c20的芳基及其衍生物、c6~c20的脂环基及其衍生物中的至少一种;和/或,在式(i-4)~式(i-6)中,r
′
选自c1~c12的烷基、c6~c12的脂环基、苯基或取代苯基;和/或,在式(i-7)~式(i-9)中,r1″
~r4″
各自独立地选自氢、c1~c12的烷基、c6~c12的脂环基或苯基。
[0018]
在进一步优选的实施方式中,在式(i-1)~式(i~9)中,每个r各自独立地选自c2~c10的烷基及其衍生物、c6~c10的芳基及其衍生物、c6~c10的脂环基及其衍生物中的至少一种;和/或,在式(i-4)~式(i-6)中,r
′
选自c1~c6的烷基、c6~c8的脂环基或苯基;和/或,在式(i-7)~式(i-9)中,r1″
~r4″
各自独立地选自氢、c1~c6的烷基、c6~c8的脂环基或苯基。
[0019]
例如,在式(i-1)~式(i-9)中,r选自:c2~c10的烷基、苯基、
[0020][0021]
在更进一步优选的实施方式中,所述封端苯并噁嗪树脂选自式(i-1)~式(i-6)所述聚合物中的至少一种:
[0022][0023][0024]
在式(i-1)~式(i-6)中,n为2~20,优选为4~14,例如为2、3、4、5、6、7、8、9、10、11、12、13、14、15、16、17、18、19或20。在式(i-1)~式(i-6)中,r、r
′
、r1″
~r4″
具有与前述相同的定义。
[0025]
本发明的目的之二在于提供一种制备本发明目的之一所述封端苯并噁嗪树脂的方法,包括:将单胺化合物和/或单酚化合物与含螺环缩醛结构的双酚类化合物、二胺类化合物、醛类化合物混合进行反应,经过后处理得到所述封端苯并噁嗪树脂。
[0026]
其中,所述方法可以采用现有技术公开的溶液法、无溶剂法和悬浮法进行,优选溶
液法。
[0027]
在一种优选的实施方式中,所述含螺环缩醛结构的双酚类化合物选自如式(ii)所示季戊四醇双缩对羟基苯甲醛(p-sq)、式(iii)所示季戊四醇双缩间羟基苯甲醛(m-sq)、式(iv)所示季戊四醇双缩邻羟基苯甲醛(o-sq)及它们的衍生物中的至少一种:
[0028][0029]
在一种优选的实施方式中,所述二胺类化合物选自脂肪族二胺及其衍生物、芳香族二胺及其衍生物、脂环族二胺及其衍生物中的至少一种。
[0030]
在进一步优选的实施方式中,所述二胺类化合物选自c2-c20的脂肪族二胺及其衍生物、c6~c20的芳基族二胺及其衍生物、c6~c20的脂环族二胺及其衍生物中的至少一种。
[0031]
在更进一步优选的实施方式中,所述二胺类化合物选自c2-c10的脂肪族二胺及其衍生物、c6~c10的芳基族二胺及其衍生物、c6~c10的脂环族二胺及其衍生物中的至少一种。
[0032]
例如,所述二胺类化合物选自乙二胺、丁二胺、己二胺、辛二胺、癸二胺、1,12-二氨基十二烷、对苯二胺、4,4'-二氨基二苯甲烷、二氨基二苯砜、4,4'-亚甲基双(2-甲基环己胺)中的至少一种。
[0033]
在一种优选的实施方式中,所述醛类化合物选自多聚甲醛和/或甲醛水溶液,优选多聚甲醛。
[0034]
其中,当选用甲醛水溶液时,其浓度为37%。
[0035]
在一种优选的实施方式中,所述含螺环缩醛结构的双酚类化合物、二胺类化合物和醛类化合物的比为1:1:(2~5)。
[0036]
在进一步优选的实施方式中,所述含螺环缩醛结构的双酚类化合物、二胺类化合物和醛类化合物的摩尔比为1:1:(2~3)。
[0037]
其中,所述含螺环缩醛结构的双酚类化合物的摩尔量以其中酚羟基的摩尔量计,所述二胺类化合物的摩尔量以其中胺基的摩尔量计,所述醛类化合物的摩尔量以其中醛基的摩尔量计。
[0038]
在一种优选的实施方式中,所述单胺化合物的结构为r
′‑
nh2。
[0039]
在进一步优选的实施方式中,所述单胺化合物选自苯胺、甲胺、乙胺、正丙胺、异丙胺、正丁胺、新戊胺、十二烷基胺和十八烷基胺中的至少一种。
[0040]
在一种优选的实施方式中,所述单酚类化合物选自苯酚、取代苯酚中的至少一种。
[0041]
在一种优选的实施方式中,所述单胺类化合物和单酚类化合物的总摩尔与含螺环缩醛结构的双酚类化合物的摩尔之比为(1~2):1,优选为(1~1.2):1,例如1:1;和/或,
[0042]
在进一步优选的实施方式中,所述单胺类化合物和所述单酚类化合物的摩尔用量比为(0~10):(10~0)。
[0043]
在一种优选的实施方式中,当将单胺化合物和/或单酚化合物与所述醛类化合物、所述含螺环缩醛结构的双酚类化合物、所述二胺类化合物一起混合进行反应时,所述反应的温度为80~150℃,优选为90~130℃;和/或,所述反应的时间为0.2~5h,优选0.5~2h。
[0044]
在一种优选的实施方式中,所述方法包括以下步骤:
[0045]
步骤1、将所述醛类化合物、所述含螺环缩醛结构的双酚类化合物、所述二胺类化合物与溶剂混合,升温进行预反应;
[0046]
步骤2、反应结束后加入单胺化合物和/或单酚化合物进行封端;
[0047]
步骤3、封端结束后进行后处理,得到所述封端苯并噁嗪树脂。
[0048]
在一种优选的实施方式中,在步骤1中,所述溶剂选自乙醇、甲醇、异丙醇、丁醇、异丁醇、四氢呋喃、二氧六环、甲苯、二甲基甲酰胺、二甲基亚砜、n-甲基吡咯烷酮中的一种或多种(例如一种或两种)。
[0049]
在进一步优选的实施方式中,在步骤1中,所述溶剂选自异丙醇、四氢呋喃、二氧六环、甲苯、二甲基甲酰胺、二甲基亚砜和n-甲基吡咯烷酮中的一种或多种(例如一种或两种)。
[0050]
在更进一步优选的实施方式中,在步骤1中,所述溶剂选自异丙醇、二氧六环、二甲基甲酰胺中的一种或多种(例如一种或两种)。
[0051]
在一种优选的实施方式中,在步骤1中,所述预反应的温度为80~150℃,优选为90~130℃,例如80℃、90℃、100℃、110℃、120℃、130℃、140℃或150℃。
[0052]
在进一步优选的实施方式中,在步骤1中,所述预反应于回流下进行。
[0053]
在一种优选的实施方式中,在步骤1中,所述预反应的时间为0.2~5h,优选0.5~2h。
[0054]
例如,在步骤1中,所述反应的时间为0.2h、0.5h、1h、1.5h、2h、2.5h、3h、3.5h、4h、4.5h或5h。
[0055]
在一种优选的实施方式中,在步骤2中,所述封端进行5~20h,优选10~12h。
[0056]
例如,在步骤2中,所述封端进行5h、6h、7h、8h、9h、10h、11h、12h、13h、14h、15h、16h、17h、18h、19h或20h。
[0057]
在一种优选的实施方式中,在步骤3中,所述后处理包括碱洗、水洗、脱溶剂处理和干燥。
[0058]
在进一步优选的实施方式中,采用碱液洗涤至中性,优选所述碱液的重量浓度为0.5~20%,优选为1~10%。
[0059]
例如,碱液的重量浓度为0.5%、1%、2%、3%、4%、5%、6%、7%、8%、9%、10%、12%、14%、16%、18%或20%。
[0060]
其中,所述碱液可以选自氢氧化钠水溶液、氢氧化钾水溶液、碳酸氢钠水溶液和碳酸氢钾水溶液等碱性水溶液中的至少一种。
[0061]
在更进一步优选的实施方式中,采用抽滤的方式脱除溶剂,然后干燥。
[0062]
本发明目的之三在于提供利用本发明目的之二所述制备方法得到的封端苯并噁嗪树脂。
[0063]
其中,所述封端苯并噁嗪树脂进行开环固化可以得到固化树脂。
[0064]
在一种优选的实施方式中,所述开环固化于100~240℃下进行,优选120~220℃。
[0065]
在进一步优选的实施方式中,所述开环固化如下进行:自120℃升温至220℃,每20℃为一个温度阶梯,每个温度阶梯反应2h。
[0066]
优选地,120℃(2h)~140℃(2h)~160℃(2h)~180℃(2h)~200℃(2h)~220℃(2h)。
[0067]
本发明所述封端苯并噁嗪树脂固化产物可以在酸性溶液中降解,优选地,苯并噁嗪树脂固化产物浸泡于酸性溶液中,反应8-48小时实现降解,其中酸性越强,时间越长,固化产物化学降解程度越高。
[0068]
优选地,所述酸性溶液中所用酸选自有机酸(例如甲酸、乙酸、三氟甲基磺酸、三氯乙酸中的至少一种)和无机酸(例如盐酸、硫酸、硝酸、磷酸中的至少一种)。
[0069]
优选地,所述酸性溶液所用溶剂为常见的极性溶剂,选自水和、醇类化合物、酮类化合物、醚类化合物、酰胺类化合物中的至少一种;更优选地,所述醇类溶剂选自乙醇、甲醇、异丙醇、丁醇、异丁醇、苯乙醇、苯甲醇、乙二醇、丁二醇、1,3-丙二醇、1,2-丙二醇、甘油、乙二醇单甲醚、二甘醇、三甘醇、二丙二醇、糠醇、四氢糠醇;所述酮类化合物选自丁酮、环己酮中的至少一种;所述醚类化合物选自thf、1,4-二氧六环;所述酰胺类化合物选自dmf、dmso、环丁砜、n-甲基吡咯烷酮、吗啉、n-甲基吗啉中的至少一种。
[0070]
本发明的目的之四在于提供一种双邻苯二甲腈基复合材料,其包括封端苯并噁嗪树脂和含螺环缩醛结构的双邻苯二甲腈类化合物,其中,所述封端苯并噁嗪树脂选自本发明目的之一所述封端苯并噁嗪树脂或利用本发明目的之二所述方法得到的主链型苯并噁嗪。
[0071]
其中,所述含螺环缩醛结构的双邻苯二甲腈类化合物中含有螺环缩醛结构,该化合物可以采用现有技术中公开的含螺环缩醛结构的双邻苯二甲腈类化合物,例如但不限于参考专利cn112010833a和文献“ke li,hua yin,kun yang,pei dai,ling han and riwei xu.synthesis and properties of phthalonitrile-based resins containing spirocycle acetal.high performance polymers 2020”,在此,将cn112010833a和该文献全文引入。
[0072]
单纯含螺环缩醛结构的双邻苯二甲腈类化合物的固化温度较高,最高固化温度高于250℃,甚至能达到300~400℃,例如,在专利cn112010833a中的实施例中最高固化温度达到375℃。但是,发明人经过大量实验发现,当将含螺环缩醛结构的双邻苯二甲腈类化合物与本发明所述主链型苯并噁嗪的混合物进行固化时,所述主链型苯并噁嗪的存在可以降低混合物中含螺环缩醛结构的双邻苯二甲腈类化合物的固化温度,具体地,该混合物在低于250℃的情况下就可以实现固化。
[0073]
另外,螺环缩醛结构在高温下(例如250℃及以上)的温度下会被破坏,当对单纯含螺环缩醛结构的双邻苯二甲腈类化合物进行固化时,由于最高固化温度明显高于250℃,这样在高温下会导致该种化合物中的螺环缩醛键被破坏,使得到的固化产物失去酸性条件下的降解性。因此,一般含螺环缩醛结构的双邻苯二甲腈类化合物的固化产物不能酸性降解。
[0074]
在本发明中,发明人经过大量实验发现,通过在含螺环缩醛结构的双邻苯二甲腈类化合物中引入本发明所述封端苯并噁嗪,降低了含螺环缩醛结构的双邻苯二甲腈类化合物的固化温度,具体地,可以在低于250℃的情况下就可以实现固化,这样,含螺环缩醛结构的双邻苯二甲腈类化合物中的螺环缩醛键不会被破坏,保留了含螺环缩醛结构的双邻苯二
甲腈类化合物的酸性可降解性。
[0075]
在一种优选的实施方式中,以所述双邻苯二甲腈基复合材料总重100wt%计,其中,封端苯并噁嗪占比10~90wt%,含螺环缩醛结构的双邻苯二甲腈类化合物占比10~90wt%。
[0076]
在进一步优选的实施方式中,以所述双邻苯二甲腈基复合材料总重100wt%计,其中,封端苯并噁嗪占比10~50wt%,含螺环缩醛结构的双邻苯二甲腈类化合物占比50~90wt%。
[0077]
例如,以所述双邻苯二甲腈基复合材料总重100wt%计,其中,封端苯并噁嗪占比10、20、30、40或50wt%,含螺环缩醛结构的双邻苯二甲腈类化合物占比50、60、70、80或90wt%。
[0078]
本发明的目的之五在于提供一种双邻苯二甲腈基复合固化树脂,其为本发明目的之四所述双邻苯二甲腈基复合材料的开环固化产物。
[0079]
在一种优选的实施方式中,所述开环固化于120~220℃下进行;优选地,自120℃升温至220℃,每10~30℃为一个温度梯度,每个温度梯度反应1~3h。
[0080]
例如,自120℃升温至220℃,每20℃为一个温度梯度,每个温度梯度反应2h,得到固化后产物。
[0081]
其中,所述双邻苯二甲腈基复合固化树脂在酸性条件下可以发生降解。
[0082]
关于含螺环缩醛结构的邻苯二甲腈类化合物固化树脂不能降解的说明:可以参见文献“ke li,hua yin,kun yang,pei dai,ling han and riwei xu.synthesis and properties of phthalonitrile-based resins containing spirocycle acetal.high performance polymers 2020”,在该文献中明确地做了实验跟分析,单纯含螺环缩醛结构的邻苯二甲腈类化合物固化树脂失去了可降解性,原因在于,高温固化过程中可能会发生螺环缩醛结构的某些反应,从而导致螺环缩醛结构的破坏形成其他结构,这可以从ftir(文献中的图8)进行解释。
[0083]
与现有技术相比,本发明具有如下有益效果:
[0084]
(1)本发明所述封端苯并噁嗪树脂中引入了缩醛结构,可以赋予树脂可降解性,利于回收再利用;(2)本发明所述封端苯并噁嗪树脂中引入的缩醛为螺环缩醛,具有一定刚性,赋予苯并噁嗪树脂固化过程较好的耐热性以及赋予固化树脂耐热性和刚性;(3)通过本发明所述封端苯并噁嗪树脂可以降低含缩醛结构的双邻苯二甲腈类化合物的固化温度;(4)本发明所述双邻苯二甲腈基复合固化树脂在酸性条件下可以发生降解。
附图说明
[0085]
图1~图12依次示出实施例1、实施例7~17得到的封端苯并噁嗪的dsc曲线。
[0086]
图13~图24依次示出实施例1、实施例7~17得到的封端苯并噁嗪的核磁谱图。
具体实施方式
[0087]
下面结合具体附图及实施例对本发明进行具体的描述,有必要在此指出的是以下实施例只用于对本发明的进一步说明,不能理解为对本发明保护范围的限制,本领域技术人员根据本发明内容对本发明做出的一些非本质的改进和调整仍属本发明的保护范围。除
非特别说明,以下实施例中使用的试剂和仪器均为市售可得产品。
[0088]
季戊四醇双缩间羟基邻苯二甲腈的制备参考专利cn112010833a或文献“ke li,hua yin,kun yang,pei dai,ling han and riwei xu.synthesis and properties of phthalonitrile-based resins containing spirocycle acetal.high performance polymers 2020”。
[0089]
实施例和对比例采用的额测试方法:
[0090]
ftir:is-5傅里叶红外光谱仪,kbr压片法;
[0091]
h-nmr:brukeravance 400mhz,25℃,氘代氯仿;本发明采用核磁结果中基团r中的h与端羟基中的h的积分面积换算得到可能的n值。
[0092]
dsc:tq100,10℃/min,氮气气氛。
[0093]
在本发明中,对于固化树脂和复合固化树脂,其降解程度(%)的计算方法如下面公式所示:
[0094]
降解程度%=[1-(w1-w2)/w1]
×
100%
[0095]
其中,w1表示固化树脂的起始重量,w2为降解及不溶性残留物重量。
[0096]
对比例1
[0097]
按照酚羟基:胺基:醛基=1:1:2(摩尔比)称量原料,依次在100ml三口烧瓶中加入3.44g(0.01mol)的季戊四醇双缩对羟基苯甲醛,0.88g(0.01mol)丁二胺,1.20g(0.04mol)多聚甲醛和50ml dmf,其中多聚甲醛1次加入;设置反应温度为115℃,反应24h。反应结束后,产物先用碳酸氢钠水溶液(0.5m)洗涤,过滤,再用去离子水洗涤至中性,过滤,真空干燥至恒重(50℃)得到(p-sq)
–
丁二胺主链型苯并噁嗪,称量,产率为54.3%。季戊四醇对羟基苯甲醛-丁二胺主链型苯并噁嗪树脂按照120,140,160,180,200,220,240℃程序升温,每个温度段保持2个小时,最后降温至室温的条件进行固化,所得固化产物用于后续化学降解的对比例;在乙醇:水:乙酸(0.1m)=4:1:1的条件下进行降解实验。
[0098][0099]
对比例2
[0100]
按照酚羟基:胺基:醛基=1:1:2.1(摩尔比)称量原料,依次在100ml三口烧瓶中加入50ml dmf,3.44g(0.01mol)季戊四醇双缩对羟基苯甲醛和1.83ml(0.02mo1)苯胺,搅拌混合均匀;向三口烧瓶中分2~4次加入1.261g(0.042mol)多聚甲醛,将三口烧瓶先置于低温恒温反应浴中搅拌30min,然后逐渐升温至95℃,恒温反应10h。反应结束后,将反应产物先用浓度为1mol/l的氢氧化钠水溶液洗涤,过滤,再用去离子水洗涤至中性,过滤,在50℃下真空干燥至恒重。
[0101]
实施例1
[0102]
按照酚羟基:胺基:醛基=1:1:2(摩尔比)称量原料,依次先在100ml三口烧瓶中加入3.44g(0.01mol)的季戊四醇双缩对羟基苯甲醛,0.44g(0.005mol)丁二胺,1.20g(0.04mol)多聚甲醛和50ml dmf,其中多聚甲醛一次加入;设置反应温度为115℃,先反应1h。再加入0.93g(0.01mol)苯胺,继续反应12h。反应结束后,产物先用碳酸氢钠水溶液
(5%)洗涤,过滤,再用去离子水洗涤至中性,再进行抽滤。将产物真空干燥至恒重(70℃)得到(p-sq)
–
丁二胺苯胺封端苯并噁嗪树脂,称量,产率为59.1%。产物分子结构如下所示。
[0103][0104]
实施例2
[0105]
按照酚羟基:胺基:醛基=1:1:2(摩尔比)称量原料,其合成过程与实施例1相同,只是溶剂选用为甲苯和乙醇的混合溶剂(甲苯:乙醇=2:1),反应温度为80℃,产率为36.7%。
[0106]
实施例3
[0107]
按照酚羟基:胺基:醛基=1:1:2(摩尔比)称量原料,其合成过程与实施例1相同,只是溶剂选用dmf和二氧六环的混合溶剂(dmf:二氧六环=2:1),产率为49.8%。
[0108]
实施例4
[0109]
按照酚羟基:胺基:醛基=1:1:2(摩尔比)称量原料,其合成过程与实施例1相同,只是溶剂选用了dmf和正丁醇的混合溶剂(dmf:正丁醇=2:1),产率为54.0%。
[0110]
实施例5
[0111]
按照酚羟基:胺基:醛基=1:1:2(摩尔比)称量原料,其合成过程与实施例1相同,只是溶剂选用了dmf和丁二醇的混合溶剂(dmf:正丁醇=2:1),产率为59.3%。
[0112]
实施例6
[0113]
合成过程与实施例1相同,只是苯胺一步加料,反应时间为12h,产率为73.6%。
[0114]
实施例7
[0115]
按照酚羟基:胺基:醛基=1:1:2(摩尔比)称量原料,依次先在100ml三口烧瓶中加入3.44g(0.01mol)的季戊四醇双缩对羟基苯甲醛,0.30g(0.005mol)乙二胺,1.20g(0.04mol)多聚甲醛和50ml dmf,其中多聚甲醛一次加入;设置反应温度为115℃,先反应1h。再加入0.93g(0.01mol)苯胺,继续反应12h。反应结束后,产物先用碳酸氢钠水溶液(5%)洗涤,过滤,再用去离子水洗涤至中性,再进行抽滤。将产物真空干燥至恒重(70℃)得到(p-sq)
–
乙二胺苯胺封端苯并噁嗪树脂,称量,产率为63.3%。产物分子结构如下所示。
[0116][0117]
实施例8
[0118]
按照酚羟基:胺基:醛基=1:1:2(摩尔比)称量原料,依次先在100ml三口烧瓶中加入3.44g(0.01mol)的季戊四醇双缩对羟基苯甲醛,0.58g(0.005mol)己二胺,1.20g(0.04mol)多聚甲醛和50ml dmf,其中多聚甲醛一次加入;设置反应温度为115℃,先反应1h。再加入0.93g(0.01mol)苯胺,继续反应12h。反应结束后,产物先用碳酸氢钠水溶液(5%)洗涤,过滤,再用去离子水洗涤至中性,再进行抽滤。将产物真空干燥至恒重(70℃)得到(p-sq)
–
己二胺苯胺封端苯并噁嗪树脂,称量,产率为53.5%。产物分子结构如下所示。
[0119]
[0120]
实施例9
[0121]
合成过程与实施例8相同,只是0.72g(0.005mol)辛二胺代替己二胺,得到(p-sq)
–
辛二胺苯胺封端苯并噁嗪树脂,产率为52.3%。产物分子结构如下所示。
[0122][0123]
实施例10
[0124]
合成过程与实施例9相同,只是0.86g(0.005mol)癸二胺代替己二胺,得到(p-sq)
–
癸二胺苯胺封端苯并噁嗪树脂,产率为50.7%。产物分子结构如下所示。
[0125][0126]
实施例11
[0127]
合成过程与实施例9相同,只是1.00g(0.005mol)1,12-二氨基十二烷代替己二胺,得到(p-sq)-1,12-二氨基十二烷苯胺封端苯并噁嗪树脂,产率为45.2%。产物分子结构如下所示。
[0128][0129]
实施例12
[0130]
合成过程与实施例9相同,只是0.98g(0.005mol)4,4'-二氨基二苯甲烷代替己二胺,得到(p-sq)-4,4
‘‑
二氨基二苯甲烷苯胺封端苯并噁嗪树脂,产率为31.1%。产物分子结构如下所示。
[0131][0132]
实施例13
[0133]
合成过程与实施例9相同,只是1.24g(0.005mol)二氨基二苯砜代替己二胺,得到(p-sq)-二氨基二苯砜苯胺封端苯并噁嗪树脂,产率30.0%。产物分子结构如下所示。
[0134][0135]
实施例14
[0136]
合成过程与实施例9相同,只是1.19g(0.005mol)4,4'-亚甲基双(2-甲基环己胺)代替己二胺,得到(p-sq)-4,4'-亚甲基双(2-甲基环己胺)苯胺封端苯并噁嗪树脂,产率为63.6%。产物分子结构如下所示。
[0137]
[0138]
实施例15
[0139]
按照酚羟基:胺基:醛基=1:1:2(摩尔比)称量原料,依次在100ml三口烧瓶中加入1.72g(0.005mol)的季戊四醇双缩对羟基苯甲醛,0.88g(0.01mol)丁二胺,1.20g(0.04mol)多聚甲醛和50ml dmf,其中多聚甲醛一次加入;设置反应温度为115℃,先反应1h。再加入0.94g(0.01mol)苯酚,继续反应12h。反应结束后,产物先用碳酸氢钠水溶液(5%)洗涤,过滤,再用去离子水洗涤至中性,再进行抽滤。将产物真空干燥至恒重(70℃),得到(p-sq)-丁二胺苯酚封端苯并噁嗪树脂,称量,产率为50.5%。产物分子结构如下所示。
[0140][0141]
实施例16
[0142]
合成过程与实施例15相同,只是1.98g(0.01mol)4,4'-二氨基二苯甲烷代替丁二胺,得到(p-sq)-4,4
‘‑
二氨基二苯甲烷苯胺封端苯并噁嗪树脂,产率33.3%。
[0143]
产物分子结构如下所示。
[0144][0145]
实施例17
[0146]
合成过程与实施例16相同,只是2.38g(0.01mol)4,4'-亚甲基双(2-甲基环己胺)代替丁二胺,得到(p-sq)-4,4'-亚甲基双(2-甲基环己胺)苯酚封端苯并噁嗪树脂,产率59.7%。产物分子结构如下所示。
[0147][0148]
实施例18
[0149]
以下以季戊四醇双缩对羟基苯甲醛-丁二胺苯胺封端苯并噁嗪树脂树脂和季戊四醇双缩对羟基苯甲醛-丁二胺苯酚封端苯并噁嗪树脂固化产物的降解为例说明含缩醛结构的苯并噁嗪树脂的降解性能。
[0150]
将季戊四醇双缩对羟基苯甲醛-丁二胺苯胺封端苯并噁嗪树脂(合成条件见实施例1)和季戊四醇双缩对羟基苯甲醛-丁二胺苯酚封端苯并噁嗪树脂(合成条件见实施例16)固化,每20℃为一个温度梯度,每个温度梯度反应2h,从120℃升温至220℃,得到固化后产物。将固化产物放入不同溶液中加热降解,降解一定时间后,过滤,干燥滤纸至恒重,测试其降解程度。降解条件及结果列在表1中。
[0151]
表1
[0152]
[0153][0154]
由上表1可以看出,采用本发明所述封端苯并噁嗪树脂固化后的固化树脂具有酸性可降解性。并且,降解温度越高、降解时间越长、降解用酸性溶液酸性越强,降解的越彻底;同时,封端苯并噁嗪的降解程度明显高于非封端苯并噁嗪。
[0155]
实施例19
[0156]
以下以季戊四醇双缩对羟基苯甲醛-丁二胺苯胺封端苯并噁嗪树脂与季戊四醇双缩间羟基邻苯二甲腈固化产物的降解为例说明含缩醛结构的噁嗪邻苯二甲腈树脂的降解性能。
[0157]
(1)将季戊四醇双缩对羟基苯甲醛-丁二胺苯胺封端苯并噁嗪树脂(合成条件见实施例1)和季戊四醇双缩对羟基邻苯二甲腈树脂按不同质量比混合后固化,每20℃为一个温度梯度,每个温度梯度反应2h,从120℃升温至220℃,得到固化后产物。将固化产物放入不同溶液中加热降解,降解一定时间后,过滤,干燥滤纸至恒重,测试其降解程度。降解条件及结果列在表2中。可以看出引入季戊四醇双缩对羟基苯甲醛-丁二胺苯胺封端苯并噁嗪后,对季戊四醇型邻苯二甲腈树脂的降解性有促进作用。其中,当噁嗪与邻苯二甲腈质量比为4:6时,固化物有较好的降解性。
[0158]
表2
[0159][0160]
(2)将季戊四醇双缩对羟基苯甲醛-丁二胺苯酚封端苯并噁嗪树脂树脂(合成条件见实施例16)和季戊四醇双缩对羟基邻苯二甲腈树脂按不同质量比混合后固化,每20℃为一个温度梯度,每个温度梯度反应2h,从120℃升温至220℃,得到固化后产物。将固化产物放入不同溶液中加热降解,降解一定时间后,过滤,干燥滤纸至恒重,测试其降解程度。降解条件及结果列在表3中。可以看出引入季戊四醇双缩对羟基苯甲醛-丁二胺苯酚封端苯并噁嗪后,对季戊四醇型邻苯二甲腈树脂的降解性有促进作用。其中,当噁嗪与邻苯二甲腈质量比为9:1时,固化物有较好的降解性,虽然与苯胺封端苯并噁嗪树脂相比降解能力有所下降,但在常温下降解能力略有提高。
[0161]
表3
[0162][0163]
实施例20热稳定性能
[0164]
对实施例和对比例2得到的树脂分别进行tga检测,结构如下表4。
[0165]
表4:
[0166]
噁嗪种类t
d5%
(℃)t
d10%
(℃)yc(780℃,%)实施例1255.69287.7746.15实施例16293.68341.4144.78对比例2230.30304.2048.36
[0167]
由表4可以看出与苯胺型噁嗪相比,本发明所述封端苯并噁嗪的初始分解温度更高、热稳定性更加优异,分析原因可能在于:由于螺环结构的刚性从而赋予苯并噁嗪较高的初始分解温度,而本发明的封端苯并噁嗪含有更多的螺环结构,因此其热稳定性更优异。
[0168]
实验例 产物检测
[0169]
(1)对对比例1得到的(p-sq)
–
丁二胺主链型苯并噁嗪进行检测:
[0170]
红外检测结果如下:1501cm-1
处为苯环上c-c的伸缩振动峰,1383cm-1
处为丁二胺中-ch
2-的吸收振动峰。1233cm-1
处为噁嗪环上c-o-c键的伸缩振动峰;1119cm-1
处是缩醛的c-o特征峰,即为缩醛环中与两个o原子相连的叔碳的c-o特征吸收峰;950cm-1
处为噁嗪环的吸收振动峰;1078cm-1
处为缩醛环上c-o-c的伸缩振动峰;1164cm-1
处也是噁嗪环上c-n-c的振动峰。可以看出,有噁嗪环存在。在2849和2937cm-1
处为缩醛环中亚甲基-ch
2-上的-ch-不对称和对称伸缩振动吸收峰。在3426cm-1
处可以观察到-oh的伸缩振动峰,表明噁嗪两端是缩醛双酚的游离羟基。
[0171]
核磁检测结果如下:在化学位移δ=5.64ppm处是质子峰ha,属于噁嗪环
–
o-ch
2-n-结构中间碳上的氢。δ=4.85ppm处出现的是质子峰hb,属于噁嗪环ar-ch
2-n-结构中间碳上的氢。因为生成了噁嗪环,所以苯环上的氢的化学位移及裂分发生了变化,质子峰hi、hj都出现在化学位移δ=6.9-7.1ppm区间内。除此以外,中间的螺环结构上的c、d、e、f四处的化学位移也从原来的4.5ppm-3.6ppm变化到了4.9ppm-3.6ppm,且有的质子峰与与hb发生了重叠。属于丁二胺上的质子峰hk出现在1.0ppm-2.7ppm区间内。进行峰面积积分,得到sa:sb:sg:s
c+d+e+f
:si:sj=2:2:1:8:1:1,其理论上各个位置氢的比例一致。因此可以确定成功合成了(p-sq)
–
丁二胺主链型苯并噁嗪。
[0172]
dsc检测,从图中可以看出第一个向下的峰是熔融吸热峰,熔融峰顶温度为150℃左右,峰很宽。第二个向上的峰则是噁嗪的固化放热峰,峰顶温度在256℃左右。
[0173]
(2)对实施例1得到的(p-sq)
–
丁二胺苯胺封端苯并噁嗪树脂进行检测:
[0174]
红外检测结果如下:1503cm-1
处的单峰是苯环上c-c的伸缩振动峰,1383cm-1
处为丁二胺的-ch
2-吸收振动峰;1234cm-1
处为噁嗪环上c-o-c键的伸缩振动峰;1119cm-1
处是缩醛的c-o特征峰,即为缩醛环中与两个o原子相连的叔碳的c-o特征吸收峰;951cm-1
处为噁嗪环的吸收振动峰;1078cm-1
处为缩醛环上c-o-c的伸缩振动峰;1164cm-1
处是噁嗪环上c-n-c的振动峰。可以看出,有噁嗪环存在。在2844和2937cm-1
处可以观察到缩醛环中亚甲基-ch
2-上的-ch-不对称和对称伸缩振动吸收峰。与未封端主链型噁嗪相比,在750和690cm-1
处出现单取代苯环的特征峰,说明苯胺已经封端。
[0175]
核磁检测结果如下:在化学位移δ=5.36ppm处新出现的质子峰hb,属于噁嗪环
–
o-ch
2-n-结构中间碳上的氢;在化学位移δ=4.63ppm处新出现的质子峰hb',属于噁嗪环ar-ch
2-n-结构中间碳上的氢;而在δ=5.46和4.67ppm处的质子峰ha和ha'分别为端基噁嗪环
–
o-ch
2-n-结构和ar-ch
2-n-结构中间碳上的氢。因为生成了噁嗪环,所以苯环上的氢的化学位移及裂分发生了变化,中间的螺环结构上的c、d、e、f四处的化学位移也从原来的4.5ppm-3.6ppm变化到了4.9ppm-3.6ppm。属于丁二胺的-ch
2-结构上氢的质子峰hg出现在1.51和2.63ppm处,因此可以确定成功合成了(p-sq)-丁二胺苯胺封端苯并噁嗪树脂。
[0176]
dsc检测,由dsc曲线中可以看出在93℃处出现噁嗪的熔融峰。在181℃处开始出现噁嗪的固化峰,峰顶温度为210℃。
[0177]
进行gpc检测,数均分子量为1390,n=1.31。
[0178]
(3)对实施例7得到的(p-sq)
–
乙二胺苯胺封端苯并噁嗪树脂进行检测
[0179]
红外检测结果如下:1501cm-1
处为苯环上c-c的伸缩振动峰,1383cm-1
处是乙二胺中-ch
2-的吸收振动峰。1119cm-1
处是缩醛的c-o特征峰,即为缩醛环中与两个o原子相连的叔碳的c-o特征吸收峰。1234cm-1
处为噁嗪环上c-o-c键的伸缩振动峰;945cm-1
处为噁嗪环
的吸收振动峰;1078cm-1
处为缩醛环上c-o-c的伸缩振动峰,1168cm-1
处是噁嗪环上c-n-c的振动峰。可以看出,有噁嗪环的存在。在2849和2956cm-1
处可观察到缩醛环中亚甲基-ch
2-上的-ch-不对称和对称伸缩振动吸收峰。与未封端主链型噁嗪相比,在750和690cm-1
处出现单取代苯环的特征峰,说明苯胺已经封端。
[0180]
核磁检测结果如下:在化学位移δ=5.41ppm处新出现的质子峰hb,属于噁嗪环
–
o-ch
2-n-结构中间碳上的氢;在化学位移δ=4.54ppm处新出现的质子峰hb',属于噁嗪环ar-ch
2-n-结构中间碳上的氢;而在δ=5.46和4.68ppm处的质子峰ha和ha'分别为端基噁嗪环
–
o-ch
2-n-结构和ar-ch
2-n-结构中间碳上的氢。除此以外,中间的螺环结构上的c、d、e、f四处的化学位移也从原来的4.5ppm-3.6ppm变化到了4.9ppm-3.6ppm。属于乙二胺上-ch
2-结构氢的质子峰hg出现在1.06和2.84ppm处。因此可以确定成功合成了(p-sq)-乙二胺苯胺封端苯并噁嗪树脂。
[0181]
dsc检测,由dsc曲线看出在117℃处有一个较宽的熔融峰。在150℃开始出现噁嗪的第一个固化峰,峰顶温度是229℃。在229℃开始出现噁嗪的第二个固化峰,峰顶温度是253℃。
[0182]
进行gpc检测,通过分子量得到n=1.2。
[0183]
(4)对实施例8得到的(p-sq)
–
己二胺苯胺封端苯并噁嗪树脂进行检测
[0184]
红外检测结果如下:1501cm-1
处为苯环上c-c的伸缩振动峰,1383cm-1
处为己二胺中-ch
2-的吸收振动峰。1233cm-1
处为噁嗪环上c-o-c键的伸缩振动峰;1115cm-1
处是缩醛的c-o特征峰,即为缩醛环中与两个o原子相连的叔碳的c-o特征吸收峰;945cm-1
处为噁嗪环的吸收振动峰;1078cm-1
处为缩醛环上c-o-c的伸缩振动峰;1164cm-1
处也是噁嗪环上c-n-c的振动峰。可以看出,有噁嗪环存在。在2856和2937cm-1
处为缩醛环中亚甲基-ch
2-上的-ch-不对称和对称伸缩振动吸收峰。与未封端主链型噁嗪相比,在750和690cm-1
处出现单取代苯环的特征峰,说明苯胺已经封端。
[0185]
核磁检测结果如下:在化学位移δ=5.39ppm处新出现的质子峰hb,属于噁嗪环
–
o-ch
2-n-结构中间碳上的氢;在化学位移δ=4.51ppm处新出现的质子峰hb',属于噁嗪环ar-ch
2-n-结构中间碳上的氢;而在δ=5.49和4.68ppm处的质子峰ha和ha'分别为端基噁嗪环
–
o-ch
2-n-结构和ar-ch
2-n-结构中间碳上的氢。除此以外,中间的螺环结构上的c、d、e、f四处的化学位移也从原来的4.5ppm-3.6ppm变化到了4.9ppm-3.6ppm。属于己二胺上-ch
2-结构的质子峰hg出现在1.23和2.68ppm处。因此可以确定成功合成了(p-sq)
–
己二胺苯胺封端苯并噁嗪树脂。
[0186]
dsc检测,由dsc曲线看出在107℃处有一个熔融峰。在240℃出现了噁嗪的固化峰,峰顶温度为255℃。
[0187]
进行gpc检测,通过分子量得到n=1.35。
[0188]
(5)对实施例9得到的(p-sq)
–
辛二胺苯胺封端苯并噁嗪树脂进行检测
[0189]
红外检测结果如下:1503cm-1
处为苯环上c-c的伸缩振动峰,1383cm-1
处为辛二胺中-ch
2-的吸收振动峰。1234cm-1
处为噁嗪环上c-o-c的伸缩振动峰;1120cm-1
处是缩醛的c-o特征峰,即为缩醛环中与两个o原子相连的叔碳的c-o特征吸收峰;930cm-1
处为噁嗪环的吸收振动峰;1078cm-1
处为缩醛上c-o-c的伸缩振动峰,1164cm-1
处是噁嗪环上c-n-c的振动峰。可以看出,有噁嗪环存在。在2925和2849cm-1
处为缩醛环中亚甲基-ch
2-上的-ch-不对称
和对称伸缩振动吸收峰。与未封端主链型噁嗪相比,在750和690cm-1
处出现单取代苯环的特征峰,说明苯胺已经封端。
[0190]
核磁检测结果如下:在化学位移δ=5.37ppm处新出现的质子峰hb,属于噁嗪环
–
o-ch
2-n-结构中间碳上的氢;在化学位移δ=4.51ppm处新出现的质子峰hb',属于噁嗪环ar-ch
2-n-结构中间碳上的氢;而在δ=5.46和4.68ppm处的质子峰ha和ha'分别为端基噁嗪环
–
o-ch
2-n-结构和ar-ch
2-n-结构中间碳上的氢。除此以外,中间的螺环结构上的c、d、e、f四处的化学位移也从原来的4.5ppm-3.6ppm变化到了4.9ppm-3.6ppm。属于辛二胺上-ch
2-结构的质子峰hg出现在1.27和2.70ppm处。因此可以确定成功合成了(p-sq)-辛二胺苯胺封端苯并噁嗪树脂。
[0191]
dsc检测,由dsc曲线可以看出在104℃处有向下一较宽的熔融峰。在244℃处出现噁嗪的固化峰,峰顶温度为250℃。
[0192]
进行gpc检测,通过分子量得到n=1.18。
[0193]
(6)对实施例10得到的(p-sq)
–
癸二胺苯胺封端苯并噁嗪树脂进行检测
[0194]
红外检测结果如下:1500cm-1
处为苯环上c-c的伸缩振动峰,1380cm-1
处为癸二胺中-ch
2-的吸收振动峰。1234cm-1
处为噁嗪环上c-o-c的伸缩振动峰;1119cm-1
处是缩醛的c-o特征峰,即为缩醛环中与两个o原子相连的叔碳的c-o特征吸收峰;945cm-1
处为噁嗪环的吸收振动峰;1078cm-1
处为缩醛上c-o-c的伸缩振动峰,1168cm-1
处是噁嗪环上c-n-c的振动峰。可以看出,有噁嗪环存在。在2931和2849cm-1
处为缩醛环中亚甲基-ch
2-上的-ch-不对称和对称伸缩振动吸收峰。与未封端主链型噁嗪相比,在750和690cm-1
处出现单取代苯环的特征峰,说明苯胺已经封端。
[0195]
核磁检测结果如下:在化学位移δ=5.39ppm处新出现的质子峰hb,属于噁嗪环
–
o-ch
2-n-结构中间碳上的氢;在化学位移δ=4.54ppm处新出现的质子峰hb',属于噁嗪环ar-ch
2-n-结构中间碳上的氢;而在δ=5.46和4.68ppm处的质子峰ha和ha'分别为端基噁嗪环
–
o-ch
2-n-结构和ar-ch
2-n-结构中间碳上的氢。除此以外,中间的螺环结构上的c、d、e、f四处的化学位移也从原来的4.5ppm-3.6ppm变化到了4.9ppm-3.6ppm。属于癸二胺上-ch
2-结构的质子峰hg出现在1.29和2.58ppm区间内。因此可以确定成功合成了(p-sq)-癸二胺苯胺封端苯并噁嗪树脂。
[0196]
dsc检测,由dsc曲线可以看出在102℃处有一向下的熔融峰。在237℃处出现噁嗪的固化峰,峰顶温度为243℃。
[0197]
进行gpc检测,通过分子量得到n=1.15。
[0198]
(7)对实施例11得到的(p-sq)-1,12-二氨基十二烷苯胺封端苯并噁嗪树脂进行检测
[0199]
红外检测结果如下:1503cm-1
处为苯环上c-c的伸缩振动峰,1383cm-1
处为1,12-二氨基十二烷二胺中-ch
2-的吸收振动峰。1234cm-1
处为噁嗪环上c-o-c的伸缩振动峰;1114cm-1
处是缩醛的c-o特征峰,即为缩醛环中与两个o原子相连的叔碳的c-o特征吸收峰;918cm-1
处为噁嗪环的吸收振动峰;1078cm-1
处为缩醛环上c-o-c的伸缩振动峰;1168cm-1
处是噁嗪环上c-n-c的振动峰。可以看出,有噁嗪环存在。2925和2849cm-1
处为缩醛环中亚甲基-ch
2-上的-ch-不对称和对称伸缩振动吸收峰。与未封端主链型噁嗪相比,在750和690cm-1
处出现单取代苯环的特征峰,说明苯胺已经封端。
ch
2-n-结构中间碳上的氢;而在δ=5.46和4.68ppm处的质子峰ha和ha'分别为端基噁嗪环
–
o-ch
2-n-结构和ar-ch
2-n-结构中间碳上的氢。除此以外,中间的螺环结构上的c、d、e、f四处的化学位移也从原来的4.5ppm-3.6ppm变化到了4.9ppm-3.6ppm,属于二氨基二苯砜上的质子峰hg和hh分别出现在6.92ppm和7.67ppm处。因此可以确定成功合成了(p-sq)-二氨基二苯砜苯胺封端苯并噁嗪树脂。
[0211]
dsc检测,由dsc图中可以看出在107℃处有一向下的熔融峰。在261℃处出现噁嗪的固化放热峰,峰顶温度为273℃。
[0212]
进行gpc检测,通过分子量得到n=1.05。
[0213]
(10)对实施例14得到的(p-sq)-4,4'-亚甲基双(2-甲基环己胺)苯胺封端苯并噁嗪树脂进行检测
[0214]
红外检测结果如下:1503cm-1
处为苯环上c-c的伸缩振动峰,1383cm-1
处为4,4'-亚甲基双(2-甲基环己胺)中-ch
2-的吸收振动峰。1459cm-1
处为胺中甲基的特征峰。1234cm-1
处为噁嗪环上c-o-c的伸缩振动峰;1114cm-1
处是缩醛的c-o特征峰,即为缩醛环中与两个o原子相连的叔碳的c-o特征吸收峰;945cm-1
处为噁嗪环的吸收振动峰;1078cm-1
处为缩醛上c-o-c的伸缩振动峰,1164cm-1
处是噁嗪环上c-n-c的振动峰。可以看出,有噁嗪环存在。在2925和2849cm-1
处为缩醛环中亚甲基-ch
2-上的-ch-不对称和对称伸缩振动吸收峰。826cm-1
处是苯环对位取代和不对称1,2,4-三取代特征峰。与未封端主链型噁嗪相比,在750和691cm-1
处出现单取代苯环的特征峰,说明苯胺已经封端。
[0215]
核磁检测结果如下:在化学位移δ=5.37ppm处新出现的质子峰hb,属于噁嗪环
–
o-ch
2-n-结构中间碳上的氢;在化学位移δ=4.51ppm处新出现的质子峰hb',属于噁嗪环ar-ch
2-n-结构中间碳上的氢;而在δ=5.46和4.68ppm处的质子峰ha和ha'分别为端基噁嗪环
–
o-ch
2-n-结构和ar-ch
2-n-结构中间碳上的氢。除此以外,中间的螺环结构上的c、d、e、f四处的化学位移也从原来的4.5ppm-3.6ppm变化到了4.9ppm-3.6ppm。6.70-7.20ppm处为苯环上的质子峰。属于4,4'-亚甲基双(2-甲基环己胺)上的亚甲基和甲基的质子峰hg和hj分别出现在0.95ppm和0.90ppm处,属于环上的质子峰hh,hi和hn则在1.20-1.70ppm处,环上的质子峰hk出现在2.99ppm处。因此可以确定成功合成了(p-sq)-4,4'-亚甲基双(2-甲基环己胺)苯胺封端苯并噁嗪树脂。
[0216]
dsc检测,从dsc曲线可以看出102℃处有一向下的熔融峰,在194℃处出现噁嗪的固化峰,峰顶温度为211℃。
[0217]
进行gpc检测,通过分子量得到n=1.08。
[0218]
(11)对实施例15得到的(p-sq)-丁二胺苯酚封端苯并噁嗪树脂进行检测
[0219]
红外检测结果如下:1501cm-1
处的单峰是苯环上c-c的伸缩振动峰,1383cm-1
处为丁二胺的-ch
2-吸收振动峰;1245cm-1
处为噁嗪环上c-o-c键的伸缩振动峰;1120cm-1
处是缩醛的c-o特征峰,即为缩醛环中与两个o原子相连的叔碳的c-o特征吸收峰;955cm-1
处为噁嗪环的吸收振动峰;1078cm-1
处为缩醛环上c-o-c的伸缩振动峰;1164cm-1
处是噁嗪环上c-n-c的振动峰。可以看出,有噁嗪环存在。在2844和2937cm-1
处可以观察到缩醛环中亚甲基-ch
2-上的-ch-不对称和对称伸缩振动吸收峰。在820cm-1
处出现的峰为苯环不对称1,2,4-三取代特征峰。与未封端主链型噁嗪相比,在750cm-1
出现苯环邻位取代的特征峰,说明苯酚已经封端。
[0220]
核磁检测结果如下:在化学位移δ=5.36ppm处新出现的质子峰hb,属于噁嗪环
–
o-ch
2-n-结构中间碳上的氢;在化学位移δ=4.51ppm处新出现的质子峰hb',属于噁嗪环ar-ch
2-n-结构中间碳上的氢;而在δ=5.37和4.54ppm处的质子峰ha和ha'分别为端基噁嗪环
–
o-ch
2-n-结构和ar-ch
2-n-结构中间碳上的氢。除此以外,中间的螺环结构上的c、d、e、f四处的化学位移也从原来的4.5ppm-3.6ppm变化到了4.9ppm-3.6ppm。6.70-7.20ppm处为苯环上的质子峰。属于丁二胺的质子峰hg出现在1.20-1.70ppm处。因此可以确定成功合成了(p-sq)-丁二胺苯酚封端苯并噁嗪树脂。
[0221]
dsc检测,由dsc曲线可以看出在96℃处有一向下较宽的熔融峰。在218℃处出现向上的噁嗪固化峰,峰顶温度为234℃。
[0222]
进行gpc检测,通过分子量得到n=1.45。
[0223]
(12)对实施例16得到的(p-sq)-4,4
‘‑
二氨基二苯甲烷苯胺封端苯并噁嗪树脂进行检测
[0224]
红外检测结果如下:1508cm-1
处为苯环上c-c的伸缩振动峰;1234cm-1
处为噁嗪环上c-o-c的伸缩振动峰;1114cm-1
处是缩醛的c-o特征峰,即为缩醛环中与两个o原子相连的叔碳的c-o特征吸收峰;945cm-1
处为噁嗪环的吸收振动峰;1078cm-1
处为缩醛环上c-o-c的伸缩振动峰;1164cm-1
处为噁嗪环上c-n-c的振动峰。810cm-1
处为对位取代苯环的峰,且峰值较高,相比于脂肪族主链型噁嗪有明显提高,因此得出噁嗪主链上含有4,4'-二氨基二苯甲烷结构。在2912和2844cm-1
处为为缩醛环中亚甲基-ch
2-上的-ch-不对称和对称伸缩振动吸收峰。在810cm-1
处出现的峰为苯环不对称1,2,4-三取代特征峰。与未封端主链型噁嗪相比,在750cm-1
出现苯环邻位取代的特征峰,说明苯酚已经封端。
[0225]
核磁检测结果如下:在化学位移δ=5.39ppm处新出现的质子峰hb,属于噁嗪环
–
o-ch
2-n-结构中间碳上的氢。此外,该处峰与端基噁嗪环
–
o-ch
2-n-结构的质子峰ha重合;在化学位移δ=4.54ppm处新出现的质子峰hb',属于噁嗪环ar-ch
2-n-结构中间碳上的氢;在4.59ppm处的质子峰ha'为端基噁嗪环ar-ch
2-n-结构中间碳上的氢。除此以外,中间的螺环结构上的c、d、e、f四处的化学位移也从原来的4.5ppm-3.6ppm变化到了4.9ppm-3.6ppm。6.70-7.20ppm处为苯环上的质子峰。属于4,4'-二氨基二苯甲烷上亚甲基的质子峰hg出现在3.83ppm处。因此可以确定成功合成了(p-sq)-4,4'-二氨基二苯甲烷苯酚封端苯并噁嗪树脂。
[0226]
dsc检测,由dsc曲线可以看出无明显熔融峰。在262℃处出现向上的噁嗪固化峰,峰顶温度为269℃。
[0227]
进行gpc检测,通过分子量得到n=1.14。
[0228]
(13)对实施例17得到的(p-sq)-4,4'-亚甲基双(2-甲基环己胺)苯酚封端苯并噁嗪树脂进行检测
[0229]
红外检测结果如下:1503cm-1
处为苯环上c-c的伸缩振动峰,1383cm-1
处为4,4'-亚甲基双(2-甲基环己胺)中-ch
2-的吸收振动峰。1460cm-1
处为胺中甲基的特征峰。1240cm-1
处为噁嗪环上c-o-c的伸缩振动峰;1119cm-1
处是缩醛的c-o特征峰,即为缩醛环中与两个o原子相连的叔碳的c-o特征吸收峰;955cm-1
处为噁嗪环的吸收振动峰;1078cm-1
处为缩醛上c-o-c的伸缩振动峰,1164cm-1
处是噁嗪环上c-n-c的振动峰。可以看出,有噁嗪环存在。在2912和2849cm-1
处为缩醛环中亚甲基-ch
2-上的-ch-不对称和对称伸缩振动吸收峰。在826cm-1
处
出现的峰为苯环不对称1,2,4-三取代特征峰。与未封端主链型噁嗪相比,在750cm-1
处出现邻位取代苯环的特征峰,说明苯酚已经封端。
[0230]
核磁检测结果如下:在化学位移δ=5.37ppm处新出现的质子峰hb,属于噁嗪环
–
o-ch
2-n-结构中间碳上的氢。此外,该处峰与端基噁嗪环
–
o-ch
2-n-结构的质子峰ha重合;在化学位移δ=4.52ppm处新出现的质子峰hb',属于噁嗪环ar-ch
2-n-结构中间碳上的氢;在4.54ppm处的质子峰ha'为端基噁嗪环ar-ch
2-n-结构中间碳上的氢。除此以外,中间的螺环结构上的c、d、e、f四处的化学位移也从原来的4.5ppm-3.6ppm变化到了4.9ppm-3.6ppm。6.70-7.20ppm处为苯环上的质子峰。属于4,4'-亚甲基双(2-甲基环己胺)上的亚甲基和甲基的质子峰hg和hj分别出现在0.94ppm和0.90ppm处,属于环上的质子峰hh,hi和hn则在1.20-1.70ppm处,环上的质子峰hk出现在2.99ppm处。因此可以确定成功合成了(p-sq)
–
4,4'-亚甲基双(2-甲基环己胺)苯酚封端苯并噁嗪树脂。
[0231]
dsc检测,由dsc曲线可以看出在105℃处有一向下很宽的熔融峰。在234℃处出现噁嗪的固化峰,峰顶温度为244℃。
[0232]
进行gpc检测,通过分子量得到n=1.09。
[0233]
以上结合具体实施方式和范例性实例对本发明进行了详细说明,不过这些说明并不能理解为对本发明的限制。本领域技术人员理解,在不偏离本发明精神和范围的情况下,可以对本发明技术方案及其实施方式进行多种等价替换、修饰或改进,这些均落入本发明的范围内。本发明的保护范围以所附权利要求为准。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1