一种二萜衍生物及其制备方法、药物组合物和应用
1.本发明属于化药领域,具体涉及一种二萜衍生物及其制备方法、药物组合物和应用。
背景技术:
2.二萜类化合物是一类结构复杂多样并具有重要生物活性的天然产物,广泛地分布在自然界、植物、动物、海洋生物中,是分子骨架由4个异戊二烯单位构成,含20个碳原子的化合物类群。结构显示多样性,主要类型有贝壳杉烷、克罗烷、松香烷、乌头烷等。许多二萜的含氧衍生物具有多方面的生物活性,有的已是重要的药物,如紫杉醇、穿心莲内酯、丹参酮、银杏内酯、雷公藤内酯、芫花酯及甜菊等都具有较强的生物活性。
3.目前没有二萜化合物衍生物抗肿瘤活性研究的报道,例如截短侧耳素在体外肿瘤细胞增殖试验中,对大多数癌细胞的杀伤能力不足,ic
50
值均大于100μm。
技术实现要素:
4.为解决前述问题,本发明提供了一种二萜衍生物,含有作为活性成分的二萜衍生物的治疗癌症药物组合物,以及制备新型二萜衍生物的方法。
5.为了实现本发明的上述目的,本发明提供如下的技术方案:
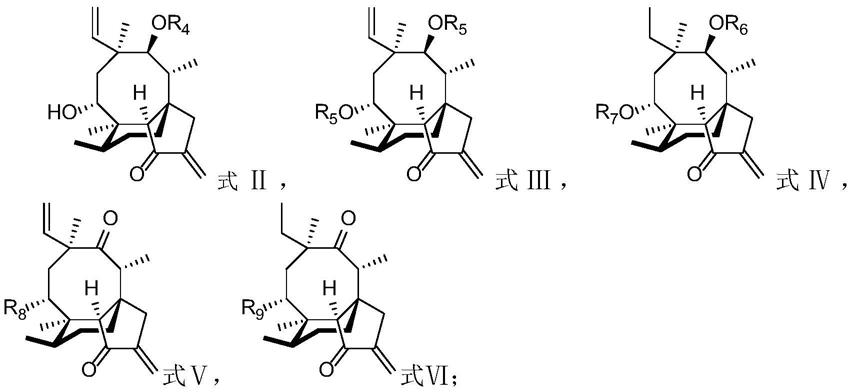
6.一种二萜衍生物,其结构如式ⅰ所示:
[0007][0008]
所述r1为氧、羟基或酯基;所述r2为氧、羟基或酯基;所述r3为乙基或乙烯基。
[0009]
优选的,上述二萜衍生物的具体结构如式ⅱ、式ⅲ、式ⅳ、式
ⅴ
或式
ⅵ
所示:
[0010][0011]
其中,r4的结构如化合物3
‑
26中的一种:
[0012]
[0013][0014]
;r5的结构如化合物27
‑
38中的一种:
[0015][0016]
;r6或r7的结构如化合物40
‑
43中的一种:
[0017][0018]
;r8的结构如化合物44
‑
45中的一种:
[0019][0020]
;r9的结构如化合物46
‑
47中的一种:
[0021][0022]
本发明的还提供了上述二萜衍生物的制备方法,其制备路线为路线一、路线二、路线三、路线四或路线五;
[0023]
路线一:
[0024][0025]
路线二:
[0026][0027]
路线三:
[0028][0029]
路线四:
[0030][0031]
路线五:
[0032][0033]
经过发明人研究发现,上述二萜衍生物能够应用在制备治疗癌症、预防癌症和/或改善癌症症状的药物或食品中。可作为治疗癌症的药物组合物的活性成分,该要用组合物还包括溶剂或可药用载体。
[0034]
上述癌症包括:妇科癌类,例如:卵巢癌、子宫颈癌、阴道癌、阴部癌、子宫/子宫内膜癌、妊娠滋养细胞肿瘤、输卵管癌、子宫肉瘤;内分泌癌类,例如:肾上腺皮质癌、脑垂体癌、胰癌、甲状腺癌、副甲状腺癌、胸腺癌、多发性内分泌肿瘤;骨癌类,例如:骨肉瘤、尤因肉瘤、软骨肉瘤等;肺癌类,例如:小细胞肺癌、非小细胞肺癌;脑和cns肿瘤,例如:神经母细胞瘤、听神经瘤、神经胶瘤和其他脑肿瘤,脊髓肿瘤、乳癌、结肠直肠癌、晚期结肠直肠腺癌;胃肠癌类,例如:肝癌、肝外胆管癌、胃肠类癌性肿瘤、胆囊癌、胃癌、食道癌、小肠癌;泌尿生殖器癌类,例如:阴茎癌、翠丸癌、前列腺癌;头和颈部肿瘤类,例如:鼻癌、鼻窦癌、鼻咽癌、口腔癌、唇癌、唾腺癌、喉头癌、下咽癌、正咽癌;血癌类,例如:急性骨髓性白血病、急性淋巴性白血病、儿童白血病、慢性淋巴性白血病、慢性骨髓性白血病、发状细胞性白血病、急性早幼粒细胞白血病、血浆细胞性白血病;骨髓癌血液病症,例如:骨髓分化不良症候群、骨髓增生性病症、范禾尼贫血、再生障碍性贫血、特发性巨球蛋白血症;淋巴癌类,例如:霍奇金病、非霍奇金氏淋巴瘤、周围t
‑
细胞林巴瘤、皮肤型t
‑
细胞淋巴瘤、aids相关性淋巴瘤;眼癌类,包括:视网膜母细胞瘤、葡萄膜黑色素瘤;皮肤癌类,例如:黑色素瘤、非黑色素瘤皮肤癌、梅克尔细胞癌;软组织肉瘤类,例如:卡波希肉瘤、儿童软组织肉瘤、成人软组织肉瘤、泌尿系统癌症,例如:肾癌维尔姆斯肿瘤、膀肤癌、尿道癌和转移性细胞癌。优选用于乳腺癌、结直肠癌、肺癌的治疗。
[0035]
本发明的有益效果为:本发明通过半合成修饰的手段获得了一种新型二萜衍生
物,制备过程简便易行,通过药理实验发现本发明提供的二萜衍生物均表现出强效的抑制多种肿瘤细胞增殖的活性,具有明显的体外和体内的抗癌活性,并且普遍不具有抗生素活性,适合用于抗肿瘤药物的开发。
附图说明
[0036]
图1所示为小鼠模型结果图,其中图1(a)为b16f10异种移植瘤体积曲线,图1(b)为定量分析肿瘤重量的情况图,图1(c)为小鼠体重定量分析情况图,图1(d)为从小鼠模型中摘除的肿瘤图像。
具体实施方式
[0037]
除有定义外,以下实施例中所用的技术术语具有与本发明创造所属领域技术人员普遍理解的相同含义。以下实施例中所用的试验试剂,如无特殊说明,均为常规生化试剂;所述实验方法,如无特殊说明,均为常规方法。
[0038]
下面结合实施例来详细说明本发明创造。
[0039]
实施例1:二萜衍生物的制备
[0040]
(1)化合物3的制备:
[0041]
化合物3的结构如下:
[0042]
其制备过程如下:
[0043]
在室温下,向截短侧耳素1(7.00g,22.1bmmol)的乙醇(50ml)和水(32ml)的溶液中加入50%氢氧化钠水溶液(3.70ml)。在65℃搅拌1小时后,将反应混合物冷却至室温并过滤,滤液用1n盐酸酸化至ph=2,然后用乙酸乙酯(200ml)稀释;分离有机相,水相用乙酸乙酯(3
×
200ml)萃取;合并的有机相用饱和食盐水洗涤,用无水硫酸钠干燥,真空浓缩,直接用于下一步。
[0044]
在室温下,向二醇化合物2(5.50克,17.2毫摩尔)的n,n
‑
二甲基甲酰胺(60毫升)溶液中分别加入四甲基甲烷二胺(60毫升)和乙酸酐(60毫升),在95℃下搅拌过夜后,反应混合物用饱和碳酸氢钠溶液(200毫升)淬灭,并用乙酸乙酯(200毫升)稀释;分离有机相,水相用乙酸乙酯(3
×
200毫升)萃取;合并的有机相用饱和食盐水洗涤,用无水硫酸钠干燥,真空浓缩,得到粗产物;然后通过硅胶快速柱色谱(石油醚∶乙酸乙酯=6∶1),得到化合物3(3.92克,68%,白色固体)。
[0045]
对化合物3进行检测,其检测结果如下:熔点142
‑
143℃,[α]
d21
=
‑
62.6
°
(c 1.45,chcl3).ir(kbr)ν
max 3563,3542,3500,2981,2929,2359,1744,1716,1455,1296,1029,996,921cm
‑1.1h nmr(400mhz,cdcl3)δ6.16(dd,j=17.8,11.1hz,1h),6.07
–
6.02(m,1h),5.41
–
5.34(m,1h),5.30(d,j=11.7hz,2h),4.41
–
4.34(m,1h),3.41(t,j=5.7hz,1h),2.32(dt,j=16.5,3.0hz,1h),2.23
–
2.15(m,3h),1.94(dd,j=15.9,7.7hz,1h),1.75(dq,j=14.4,2.8hz,1h),1.68
–
1.56(m,3h),1.49(dd,j=13.3,3.0hz,1h),1.44(s,3h),1.36(ddd,j=
21.6,8.9,4.6hz,2h),1.15(s,3h),1.06(dd,j=14.0,4.4hz,1h),0.98(d,j=7.0hz,3h),0.94(d,j=7.1hz,3h).
13
c nmr(100mhz,cdcl3)δ205.5,143.4,139.4,118.3,115.9,75.1,66.7,59.6,45.3,45.1,43.1,42.8,37.0,36.1,34.8,31.5,28.6,27.1,18.2,13.6,11.5.hrms(esi):m/zcalcd for c
21
h
31
o3‑
[m
‑
h]
‑
:331.2279;found 331.2276。
[0046]
(2)化合物4的制备:
[0047]
化合物4的结构如下:
[0048]
其制备过程如下:
[0049]
在0℃下,向化合物3(200毫克,0.602毫摩尔)的二氯甲烷(2毫升)溶液中加入三乙胺(0.125毫升,0.903毫摩尔)和dmap(7.33毫克,0.0602毫摩尔)。搅拌10分钟后,加入乙酸酐(68.0微升,0.722毫摩尔)。搅拌1小时后,所得混合物用水(3毫升)淬灭,并用二氯甲烷(3毫升)稀释。分离有机相,水相用二氯甲烷(3
×
5毫升)萃取。合并的有机相用无水硫酸钠干燥,真空浓缩,得到粗产物,通过硅胶快速柱色谱纯化(pe∶ea=12∶1),得到化合物4(126毫克,56%,白色固体)。
[0050]
对化合物4进行检测,其结果如下:熔点152
‑
153℃,[α]
d21
=
‑
49.8
°
(c 1.24,ch cl3).ir(kbr)ν
max 3588,2991,2878,1712,1664,1358,1330,1240,1025,965c m
‑1.1h nmr(400mhz,cdcl3)δ6.03(dd,j=17.8,11.1hz,1h),5.91(s,1h),5.25
–
5.08(m,3h),4.46(d,j=8.8hz,1h),3.08(d,j=18.2hz,1h),2.24
–
2.07(m,3h),2.02(s,3h),1.93(d,j=13.5hz,1h),1.77(dt,j=13.3,2.8hz,1h),1.73
–
1.65(m,2h),1.39(d,j=3.9hz,1h),1.35(s,3h),1.29(d,j=12.1hz,1h),1.15
–
0.99(m,2h),0.95(d,j=6.5hz,3h),0.86(s,3h),0.76(d,j=7.1hz,3h).
13
c nm r(100mhz,cdcl3)δ203.5,170.6,146.8,141.9,114.3,113.5,79.2,49.5,47.7,46.5,44.3,39.4,37.6,37.1,34.9,33.9,27.9,27.1,27.0,20.7,19.4,12.8,12.3.hr ms(esi):m/zcalcd for c
23
h
34
nao
4+
[m+na]
+
:397.2349;found 397.2350。
[0051]
(3)化合物5的制备:
[0052]
化合物5的结构如下:
[0053]
其制备过程如下:
[0054]
在0℃下,向化合物3(200毫克,0.602毫摩尔)的二氯甲烷(2毫升)溶液中加入n,n
‑
二异丙基乙胺(0.124毫升,0.722毫摩尔)。搅拌10分钟后,加入丙酰氯(58.0微升,0.662毫摩尔)。搅拌1小时后,所得混合物用水(10毫升)淬灭,并用二氯甲烷(10毫升)稀释。分离有机相,水相用二氯甲烷(3
×
10毫升)萃取。合并的有机相用无水硫酸钠干燥,真空浓缩,得到粗产物,用硅胶快速柱色谱纯化(pe/ea=12:1),得到化合物5(96.1毫克,41%,白色固体)。
[0055]
对化合物5进行检测,其检测结果如下:熔点156
‑
157℃,[α]
d21
=
‑
46.6
°
(c 1.28,
chcl3).ir(kbr)ν
max 3582,2987,2877,1718,1701,1640,1293,1223,1106,934cm
‑1.1h nmr(400mhz,cdcl3)δ6.10(dd,j=17.9,11.2hz,1h),6.04
–
5.98(m,1h),5.43(d,j=17.9hz,1h),5.27(d,j=11.2hz,2h),4.82(d,j=6.6hz,1h),4.32(d,j=7.2hz,1h),2.68(d,j=16.6hz,1h),2.39(qd,j=7.5,1.5hz,2h),2.32
–
2.20(m,2h),2.07(d,j=16.6hz,1h),1.96(dd,j=15.9,7.4hz,1h),1.76
–
1.61(m,3h),1.45(d,j=1.5hz,4h),1.41
–
1.30(m,2h),1.27
–
1.21(m,1h),1.17(td,j=7.6,1.6hz,3h),1.07(td,j=14.0,4.2hz,1h),0.97(dd,j=4.8,1.8h z,6h),0.78(d,j=7.0hz,3h).
13
c nmr(100mhz,cdcl3)δ205.4,173.8,143.5,140.0,118.2,115.6,76.6,66.9,59.5,44.6,44.2,42.9,42.7,36.9,36.4,34.9,31.5,29.0,27.6,27.2,18.2,13.6,12.0,9.3.hrms(esi):m/zcalcd for c
24
h
36
nao4[m+na]
+
:411.2506;found 411.2508。
[0056]
(4)化合物6的制备:
[0057]
化合物6的结构如下:
[0058]
其制备过程如下:
[0059]
向环丁烷羧酸(120毫克,1.20毫摩尔)、n,n'
‑
二异丙基碳二亚胺(0.152毫升,0.963毫摩尔)和4
‑
二甲氨基吡啶(22.4毫克,0.181毫摩尔)在n,n
‑
二甲基甲酰胺(2毫升)中的溶液中加入化合物3(200毫克,0.602毫摩尔)。在40℃搅拌过夜后,所得混合物用水(3毫升)淬灭,并用乙酸乙酯(3毫升)稀释。分离有机相,水相用乙酸乙酯(3
×
5毫升)萃取。合并的有机相用饱和食盐水洗涤,用无水硫酸钠干燥,真空浓缩,得到粗产物,用硅胶快速柱色谱纯化(pe∶ea=12∶1),得到化合物6(152毫克,61%,白色固体)。
[0060]
对化合物6进行检测,其检测结果如下:熔点190
‑
192,℃[α]
d22
=
‑
42.9
°
(c 1.53,chcl3).ir(kbr)ν
max 3554,2991,2953,2864,1718,1699,1402,1383,1357,1341,1027,923,892cm
‑1.1h nmr(400mhz,cdcl3)δ6.11(ddd,j=18.0,11.2,1.1hz,1h),6.05
–
6.01(m,1h),5.44(dd,j=17.9,1.3hz,1h),5.29
–
5.25(m,2h),4.82(d,j=6.7hz,1h),4.33(t,j=6.4hz,1h),3.20(pd,j=8.6,1.1hz,1h),2.71(dt,j=16.6,3.3hz,1h),2.36
–
2.21(m,6h),2.08(dt,j=16.5,1.7hz,1h),2.04
–
1.95(m,2h),1.93(dd,j=5.1,4.0hz,1h),1.73
–
1.63(m,3h),1.58(s,1h),1.46(s,3h),1.34(dd,j=20.9,4.5hz,2h),1.12
–
1.04(m,1h),0.97(d,j=7.0hz,6h),0.78(d,j=7.1hz,3h).
13
c nmr(100mhz,cdcl3)δ205.5,174.8,143.7,140.2,118.3,115.7,76.6,67.1,59.7,44.8,44.4,43.1,42.9,38.5,37.0,36.6,35.1,31.6,29.1,27.4,25.6,25.5,18.8,18.3,13.7,12.1.hr ms(esi):m/zcalcd for c
26
h
38
nao
4+
[m+na]
+
:437.2662;found 437.2669。
[0061]
(5)化合物7的制备:
[0062]
化合物7的结构如下:
[0063]
其制备过程如下:
[0064]
使用环戊烷羧酸(137毫克,1.20毫摩尔),按照化合物6所述的合成步骤获得目标化合物7。快速柱色谱(石油醚∶乙酸乙酯=12∶1)得到化合物7(100毫克,39%,白色固体)。
[0065]
(6)化合物8的制备:
[0066]
化合物8的结构如下:
[0067]
其制备过程如下:
[0068]
使用6
‑
溴己酸(234毫克,1.20毫摩尔),按照化合物6所述的合成步骤获得目标化合物8。快速柱色谱(石油醚∶乙酸乙酯=12∶1)得到化合物8(73.3毫克,24%,白色固体)。
[0069]
(7)化合物9的制备:
[0070]
化合物9的结构如下:
[0071]
其制备过程如下:
[0072]
使用8
‑
溴辛酸(268毫克,1.20毫摩尔),按照化合物6所述的合成步骤获得目标化合物9。快速柱色谱(石油醚∶乙酸乙酯=12∶1)得到化合物9(103毫克,32%,白色固体)。
[0073]
(8)化合物10的制备:
[0074]
化合物10的结构如下:
[0075]
其制备过程如下:
[0076]
使用焦碳酸二叔丁酯(157毫克,1.20毫摩尔),按照化合物4所述的合成步骤获得目标化合物10。快速柱色谱(石油醚∶乙酸乙酯=12∶1)得到化合物10(150毫克,58%,白色固体)。
[0077]
(9)化合物11的制备:
[0078]
化合物11的结构如下:
[0079]
其制备过程如下:
[0080]
使用碳酸丙酯(81.1毫克,0.662毫摩尔),按照化合物5所述的步骤获得目标化合物11。快速柱色谱(石油醚∶乙酸乙酯=12∶1)得到化合物11(146毫克,58%,白色固体)。
[0081]
(10)化合物12的制备:
[0082]
化合物12的结构如下:
[0083]
其制备过程如下:
[0084]
使用碳酸异丁酯(90.0毫克,0.662毫摩尔),按照化合物5所述的步骤获得目标化合物12。快速柱色谱(石油醚∶乙酸乙酯=12∶1)得到化合物12(125毫克,48%,白色固体)。
[0085]
(11)化合物13的制备:
[0086]
化合物13的结构如下:
[0087]
其制备过程如下:
[0088]
使用苯甲酸(146毫克,1.20毫摩尔),按照化合物6所述的步骤获得目标化合物13。快速柱色谱(石油醚∶乙酸乙酯=12∶1)得到化合物13(68.3毫克,26%,白色固体)。
[0089]
(12)化合物14的制备:
[0090]
化合物14的结构如下:
[0091]
其制备过程如下:
[0092]
使用4
‑
氟苯甲酸(168毫克,1.20毫摩尔),按照化合物6所述的合成步骤得到化合物14。快速柱色谱(聚乙烯∶乙酸乙酯=12∶1)得到14(109毫克,40%,白色固体)。
[0093]
(13)化合物15的制备:
[0094]
化合物15的结构如下:
[0095]
其制备过程如下:
[0096]
使用3
‑
氟苯甲酸(168毫克,1.20毫摩尔),按照化合物6所述的合成步骤获得目标化合物15。快速柱色谱(石油醚∶乙酸乙酯=12∶1)得到化合物15(161毫克,59%,白色固体)。
[0097]
对化合物15进行检测,其检测结果如下:熔点233
‑
234℃。[α]
d21
=
‑
38.6
°
(c 1.45,chcl3).ir(kbr)ν
max 3558,2981,2922,1716,1655,1538,1464,1129,974,758cm
‑1.1h nmr(400mhz,cdcl3)δ7.86(d,j=7.8hz,1h),7.73(ddd,j=9.3,2.7,1.5hz,1h),7.46(td,j=8.0,5.6hz,1h),7.30(td,j=8.3,2.0hz,1h),6.26(dd,j=17.9,11.2hz,1h),6.07(s,1h),5.52(dd,j=17.9,1.1hz,1h),5.37(d,j=11.3hz,1h),5.30(s,1h),5.09(d,j=6.8hz,1h),4.40(t,j=6.4hz,1h),2.77(dt,j=16.5,3.0hz,1h),2.42(p,j=7.0hz,1h),2.35(d,j=2.4hz,1h),2.12(d,j=16.5hz,1h),2.05(dd,j=16.0,7.5hz,1h),1.75(d,j=16.1hz,1h),1.72
–
1.55(m,3h),1.49(s,3h),1.38(dd,j=13.8,3.4hz,2h),1.11(td,j=14.0,4.3hz,1h),1.02(s,3h),1.00(d,j=7.1hz,3h),0.84(d,j=7.1hz,3h).
13
c nmr(100mhz,cdcl3)δ205.4,163.2(d,j
f
‑
c
=335.6hz),143.5,139.9,132.2(d,j
f
‑
c
=7.5hz),130.4(d,j
f
‑
c
=7.6hz),125.6,125.6,120.5(d,j
f
‑
c
=21.7hz),118.5,116.7(d,j
f
‑
c
=22.9hz),116.2,78.2,67.1,59.8,45.0,44.5,43.2,42.9,37.0,36.8,35.1,31.6,29.1,27.3,18.3,13.7,12.4.
19
f nmr(376mhz,cdcl3)δ
‑
62.7.hrms(esi):m/zcalcd for c
28
h
35
fnao4[m+na]
+
:477.2412;found 477.2418。
[0098]
(14)化合物16的制备:
[0099]
化合物16的结构如下:
[0100]
其制备过程如下:
[0101]
使用2
‑
氟苯甲酸(168毫克,1.20毫摩尔),按照6所述的步骤获得目标化合物16。快速柱色谱(石油醚∶乙酸乙酯=12∶1)得到化合物16(139毫克,51%,白色固体)。
[0102]
(15)化合物17的制备:
[0103]
化合物17的结构如下:
[0104]
其制备过程如下:
[0105]
使用4
‑
(三氟甲基)苯甲酸(228毫克,1.20毫摩尔),按照6所述的步骤获得目标化合物17。快速柱色谱(石油醚∶乙酸乙酯=12∶1)得到化合物17(154毫克,51%,白色固体)。
[0106]
对化合物17进行检测,其检测结果如下:熔点175
‑
177℃。[α]
d21
=
‑
37.6
°
(c 1.19,chcl3).ir(kbr)ν
max 2985,2935,2863,1721,1645,1326,1311,1280,1170,1132,1101,862,773,704cm
‑1.1h nmr(400mhz,cdcl3)δ8.17(d,j=7.8h z,2h),7.73(d,j=7.8hz,2h),6.26(dd,j=17.6,11.3hz,1h),6.06(s,1h),5.52(d,j=17.8hz,1h),5.37(d,j=11.2hz,1h),5.29(s,1h),5.10(d,j=6.2hz,1h),4.39(d,j=5.9hz,1h),2.75(d,j=16.1hz,1h),2.49
–
2.38(m,1h),2.34(s,1h),2.11(d,j=16.8hz,1h),2.09
–
1.98(m,1h),1.81
–
1.62(m,3h),1.54
–
1.44(m,5h),1.37(d,j=13.7hz,1h),1.10(td,j=16.7,15.5,7.6hz,1h),1.01(s,3h),0.98(d,j=6.7hz,3h),0.83(d,j=6.4hz,3h).
13
c nmr(100mhz,cdcl3)δ205.2,164.7,143.3,139.6,134.7(q,j
f
‑
c
=32.5hz),133.0,130.1,125.7(d,j
f
‑
c
=3.6hz),123.6(d,j
f
‑
c
=274.1hz),118.4,116.2,78.3,66.9,59.6,44.8,44.4,43.0,42.8,36.9,36.6,35.0,31.5,28.9,27.2,18.2,13.6,12.3.
19
f nmr(376mhz,cdcl3)δ
‑
63.1.hrms(esi):m/zcalcd for c
29
h
35
f3nao4[m+na]
+
:527.2380;found 527.2384。
[0107]
(16)化合物18的制备:
[0108]
化合物18的结构如下:
[0109]
其制备过程如下:
[0110]
使用3
‑
(三氟甲基)苯甲酸(228毫克,1.20毫摩尔),按照化合物6所述的合成步骤获得目标化合物18。快速柱色谱(石油醚∶乙酸乙酯=12∶1)得到化合物18(124毫克,41%,白色固体)。
[0111]
(17)化合物19的制备:
[0112]
化合物19的结构如下:
[0113]
其制备过程如下:
[0114]
使用茴香酸(186毫克,1.20毫摩尔),按照化合物6所述的合成步骤获得目标化合物19。快速柱色谱(石油醚∶乙酸乙酯=12∶1)获得化合物19(84.4毫克,30%,白色固体)。
[0115]
(18)化合物20的制备:
[0116]
化合物20的结构如下:
[0117]
其制备过程如下:
[0118]
使用3
‑
苯氧基苯甲酸(257毫克,1.20毫摩尔),按照化合物6所述的合成步骤获得目标化合物20。快速柱色谱(石油醚∶乙酸乙酯=12∶1)得到化合物20(152毫克,48%,白色固体)。
[0119]
(19)化合物21的制备:
[0120]
化合物21的结构如下:
[0121]
其制备过程如下:
[0122]
使用对硝基苯甲酸(200毫克,1.20毫摩尔),按照化合物6所述的合成步骤获得目标化合物21。快速柱色谱(石油醚∶乙酸乙酯=12∶1)得到化合物21(162毫克,78%,白色固体)。
[0123]
(20)化合物22的制备:
[0124]
化合物22的结构如下:
[0125]
其制备过程如下:
[0126]
使用4
‑
溴苯甲酸(241毫克,1.20毫摩尔),按照化合物6所述的合成步骤获得目标化合物22。快速柱色谱(石油醚∶乙酸乙酯=12∶1)获得化合物22(178毫克,56%,白色固体)。
[0127]
对化合物22进行检测,其检测结果如下:熔点143
‑
145℃,[α]
d22
=
‑
44.6
°
(c 1.28,chcl3).ir(kbr)ν
max 3582,2980,2901,1720,1663,1431,1330,1301,1185,979cm
‑1.1h nmr(400mhz,cdcl3)δ7.82(d,j=8.3hz,1h),7.48
–
7.39(m,2h),6.21(dd,j=17.9,11.2hz,1h),6.09
–
6.03(m,1h),5.48(d,j=17.8hz,1h),5.35
–
5.28(m,2h),5.09(d,j=6.7hz,1h),4.38(t,j=6.1hz,1h),2.79(d,j=3.3hz,1h),2.60(s,3h),2.43
–
2.34(m,2h),2.11(dt,j=16.6,1.9hz,1h),2.07
–
2.00(m,1h),1.72(s,3h),1.56
–
1.49(m,1h),1.47(s,3h),1.43
–
1.33(m,2h),1.12(dd,j=14.2,4.6hz,1h),1.01(s,3h),0.99(d,j=7.0hz,3h),0.85(d,j=7.0hz,3h).
13
c nmr(100mhz,cdcl3)δ205.4,165.9,143.5,143.1,140.0,134.9,131.9,129.3,128.0,127.1,118.4,116.1,77.5,67.0,59.7,44.9,44.5,43.1,42.8,37.0,36.7,
35.1,31.6,29.1,27.3,22.0,18.3,13.7,12.6.h rms(esi):m/zcalcd for c
27
h
4179
brnao
4+
[m+na]
+
:531.2080;found 531.2085。
[0128]
(21)化合物23的制备:
[0129]
化合物23的结构如下:
[0130]
其制备过程如下:
[0131]
使用3
‑
溴
‑2‑
甲基苯甲酸(258毫克,1.20毫摩尔),按照6所述的步骤获得目标化合物23。快速柱色谱(石油醚∶乙酸乙酯=12∶1)获得化合物23(200毫克,63%,白色固体)。
[0132]
(22)化合物24的制备:
[0133]
化合物24的结构如下:
[0134]
其制备过程如下:
[0135]
使用5
‑
三氟甲基吡啶
‑2‑
羧酸(229毫克,1.20毫摩尔),按照6所述的步骤获得目标化合物24。快速柱色谱(石油醚∶乙酸乙酯=12∶1)获得化合物24(200毫克,63%,白色固体)。
[0136]
(23)化合物25的制备:
[0137]
化合物25的结构如下:
[0138]
其制备过程如下:
[0139]
使用4
‑
氟苯乙酸(185毫克,1.20毫摩尔),按照化合物6所述的合成步骤获得目标化合物25。快速柱色谱(石油醚∶乙酸乙酯=12∶1)获得化合物25(152毫克,54%,白色固体)。
[0140]
(24)化合物26的制备:
[0141]
化合物26的结构如下:
[0142]
其制备过程如下:
[0143]
使用三氟乙酸酐(151毫克,0.722毫摩尔),按照化合物4所述的合成步骤获得目标化合物26。快速柱色谱(石油醚∶乙酸乙酯=12∶1)获得化合物26(137毫克,53%,白色固体)。
[0144]
(25)化合物27的制备:
[0145]
化合物27的结构如下:
[0146]
其制备过程如下:
[0147]
在0℃下,向α
‑
亚甲基羰基3(200毫克,0.602毫摩尔)的二氯甲烷(2毫升)溶液中加入三乙胺(0.248毫升,1.81毫摩尔)和4
‑
二甲氨基吡啶(14.6毫克,0.120毫摩尔)。搅拌10分钟后,加入乙酸酐(0.142毫升,1.44毫摩尔)。搅拌1小时后,所得混合物用水(3毫升)淬灭,并用二氯甲烷(3毫升)稀释。分离有机相,水相用二氯甲烷(3
×
5ml)萃取。合并的有机相用无水硫酸钠干燥,真空浓缩,得到粗产物。通过硅胶快速柱色谱纯化(石油醚∶乙酸乙酯=18∶1),得到化合物27(223毫克,89%,白色固体)。
[0148]
(26)化合物28的制备:
[0149]
化合物28的结构如下:
[0150]
其制备过程如下:
[0151]
向环丁烷羧酸(240毫克,2.40毫摩尔)、三乙胺(0.335毫升,1.93毫摩尔)和4
‑
二甲氨基吡啶(44.2毫克,0.362毫摩尔)的n,n
‑
二甲基甲酰胺(2毫升)溶液中加入α
‑
亚甲基羰基化合物3(200毫克,0.602毫摩尔)。在40℃搅拌过夜后,所得混合物用水(3毫升)淬灭,并用乙酸乙酯(3毫升)稀释。分离有机相,水相用乙酸乙酯(3
×
5毫升)萃取。合并的有机相用饱和食盐水洗涤,用无水硫酸钠干燥,真空浓缩,得到粗产物。通过硅胶快速柱色谱纯化(石油醚/乙酸乙酯=12∶1),得到28(225毫克,76%,白色固体)。
[0152]
(27)化合物29的制备:
[0153]
化合物29的结构如下:
[0154]
其制备过程如下:
[0155]
使用6
‑
溴己酸(468毫克,2.40毫摩尔),按照化合物28所述的合成步骤获得目标化合物29。快速柱色谱(石油醚∶乙酸乙酯=18∶1)得到化合物29(201毫克,50%,白色固体)。
[0156]
(28)化合物30的制备:
[0157]
化合物30的结构如下:
[0158]
其制备过程如下:
[0159]
使用8
‑
溴辛酸(535毫克,2.40毫摩尔),按照化合物28所述的步骤获得目标化合物30。快速柱色谱(石油醚∶乙酸乙酯=18∶1)得到30(187毫克,42%,白色固体)。
[0160]
(29)化合物31的制备:
[0161]
化合物31的结构如下:
[0162]
其制备过程如下:
[0163]
使用环丙烷羧酸(207毫克,2.40毫摩尔),按照化合物28所述的步骤获得目标化合物31。快速柱色谱(石油醚∶乙酸乙酯=18∶1)获得目标化合物31(149毫克,53%,白色固体)。
[0164]
(30)化合物32的制备:
[0165]
化合物32的结构如下:
[0166]
其制备过程如下:
[0167]
使用环丁烷羧酸(240毫克,2.40毫摩尔),按照化合物28所述的合成步骤获得目标化合物32。快速柱色谱(石油醚∶乙酸乙酯=18∶1)得到化合物32(125毫克,42%,白色固体)。
[0168]
(31)化合物33的制备:
[0169]
化合物33的结构如下:
[0170]
其制备过程如下:
[0171]
使用环戊烷羧酸(274毫克,2.40毫摩尔),按照28所述的步骤获得目标化合物33。快速柱色谱(石油醚∶乙酸乙酯=18∶1)得到化合物33(139毫克,44%,白色固体)。
[0172]
(32)化合物34的制备:
[0173]
化合物34的结构如下:
[0174]
其制备过程如下:
[0175]
使用4
‑
(三氟甲基)苯甲酸(456毫克,2.40毫摩尔),按照化合物28所述的步骤获得目标化合物34。快速柱色谱(石油醚∶乙酸乙酯=18∶1)得到化合物34(240毫克,59%,白色固体)。
[0176]
(33)化合物35的制备:
[0177]
化合物35的结构如下:
[0178]
其制备过程如下:
[0179]
使用3
‑
氟苯甲酸(336毫克,2.40毫摩尔),按照化合物28所述的步骤获得目标化合物35。快速柱色谱(石油醚∶乙酸乙酯=18∶1)获得化合物35(173毫克,50%,白色固体)。
[0180]
(34)化合物36的制备:
[0181]
化合物36的结构如下:
[0182]
其制备过程如下:
[0183]
使用3
‑
苯氧基苯甲酸(514毫克,2.40毫摩尔),按照28所述的步骤获得目标化合物36。快速柱色谱(石油醚∶乙酸乙酯=18∶1)得到化合物36(222毫克,51%,白色固体)。
[0184]
(35)化合物37的制备:
[0185]
化合物37的结构如下:
[0186]
其制备过程如下:
[0187]
使用2
‑
氯
‑6‑
甲基异烟酸(412毫克,2.40毫摩尔),按照化合物28所述的合成步骤获得目标化合物37。快速柱色谱(石油醚∶乙酸乙酯=18∶1)得到化合物37(165毫克,43%白色固体)。
[0188]
(36)化合物38的制备:
[0189]
化合物38的结构如下:
[0190]
其制备过程如下:
[0191]
使用5
‑
(三氟甲基)吡啶甲酸(459毫克,2.40毫摩尔),按照28所述的步骤获得目标化合物38。快速柱色谱(石油醚∶乙酸乙酯=18∶1)得到化合物38(248毫克,61%,白色固体)。
[0192]
对化合物38进行检测,其检测结果如下:熔点204
‑
205℃。[α]
d21
=
‑
18.8
°
(c 1.45,chcl3).ir(kbr)ν
max 2989,2964,1722,1331,1310,1170,1140,1077,1016,941,853,706cm
‑1.1h nmr(400mhz,cdcl3d)δ9.04(d,j=8.1hz,2h),8.19(d,j=8.0hz,2h),8.16
–
8.07(m,2h),6.59(dd,j=17.5,11.2hz,1h),6.08(s,2h),5.46
–
5.32(m,2h),5.31(s,1h),5.25(d,j=6.7hz,1h),2.85(d,j=16.6hz,1h),2.76(p,j=6.9hz,1h),2.54
–
2.49(m,1h),2.38(dd,j=16.1,8.3hz,1h),2.15(d,j=16.6hz,1h),1.84
–
1.57(m,7h),1.39(d,j=12.5hz,1h),1.20
–
1.03(m,4h),0.90(d,j=6.9hz,3h),0.69(d,j=6.4hz,3h).
13
c nmr(100mhz,cdcl3)δ204.5,163.2,162.6,151.4,150.6,147.3(d,j
f
‑
c
=4.6hz),147.1(d,j
f
‑
c
=4.0h z),143.2,139.3,136.1(d,j
f
‑
c
=120.2hz),134.8(dd,j
f
‑
c
=6.7,3.5hz),130.0(d,j
f
‑
c
=37.2hz),129.6(q,j
f
‑
c
=32.9hz),129.4(q,j
f
‑
c
=33.4hz),124.7,124.7,123.0(d,j
f
‑
c
=273.0hz),119.0,117.1,79.2,70.7,58.9,45.3,43.4,43.3,42.9,36.9,36.1,35.0,31.5,27.4,27.0,16.7,15.3,12.3.
19
f nmr(376mhz,cdcl3)δ
‑
62.7,
‑
62.7.hrms(esi):m/zcalcd for c
35
h
36
f6n2nao
5+
[m+na]
+
:701.2421;found 701.2425。
[0193]
(37)化合物40的制备:
[0194]
化合物40的结构如下:
[0195]
其制备过程如下:
[0196]
使用化合物39(2.00克,6.32毫摩尔),按照化合物3所述的合成步骤获得目标化合物40。快速柱色谱(石油醚∶乙酸乙酯=6∶1)得到化合物40(1.14克,56%,白色固体)。
[0197]
(38)化合物41的制备:
[0198]
化合物41的结构如下:
[0199]
其制备过程如下:
[0200]
使用乙酸酐(68.0微升,0.722毫摩尔),按照化合物4所述的步骤获得目标化合物41。快速柱色谱(石油醚∶乙酸乙酯=12∶1)获得化合物41(133毫克,59%,白色固体)。
[0201]
(39)化合物42的制备:
[0202]
化合物42的结构如下:
[0203]
其制备过程如下:
[0204]
使用乙酸酐(0.136毫升,1.44毫摩尔),按照化合物27所述的步骤获得目标化合物42。快速柱色谱(石油醚∶乙酸乙酯=18∶1)得到42(173毫克,69%,白色固体)。
[0205]
(40)化合物43的制备:
[0206]
化合物43的结构如下:
[0207]
其制备过程如下:
[0208]
使用5
‑
(三氟甲基)吡啶甲酸(459毫克,2.40毫摩尔),按照化合物28所述的合成步骤获得目标化合物43。快速柱色谱(聚乙烯∶乙酸乙酯=18∶1)得到43(236毫克,58%,白色固体)。
[0209]
(41)化合物44的制备:
[0210]
化合物44的结构如下:
[0211]
其制备过程如下:
[0212]
在室温下,向化合物3(200毫克,0.602毫摩尔)的二氯甲烷(5毫升)溶液中加入德斯
‑
马丁氧化剂(305毫克,0.662毫摩尔)。搅拌1小时后,用30毫升饱和硫代硫酸钠溶液和30毫升饱和碳酸氢钠溶液稀释反应液,所得混合物用二氯甲烷(20毫升)稀释。分离有机相,水相用二氯甲烷(3
×
5毫升)萃取。合并的有机相用无水硫酸钠干燥,真空浓缩,得到粗产物,通过硅胶快速柱色谱纯化(pe∶ea=12∶1),得到化合物44(169毫克,85%,白色固体)。
[0213]
(42)化合物45的制备:
[0214]
化合物45的结构如下:
[0215]
其制备过程如下:
[0216]
在室温下,向二醇化合物3(200毫克,0.602毫摩尔)的二氯甲烷(5毫升)溶液中加入戴斯
‑
马丁氧化剂(560毫克,1.32毫摩尔)。搅拌1小时后,用30毫升饱和硫代硫酸钠溶液和30毫升饱和碳酸氢钠溶液稀释反应液,所得混合物用二氯甲烷(20毫升)稀释。分离有机
相,水相用二氯甲烷(3
×
5毫升)萃取。合并的有机相用无水硫酸钠干燥,真空浓缩,得到粗产物,通过硅胶快速柱色谱纯化(pe∶ea=12∶1),得到化合物45(158毫克,80%,白色固体)。
[0217]
(43)化合物46的制备:
[0218]
化合物46的结构如下:
[0219]
其制备过程如下:
[0220]
使用40(200毫克,0.602毫摩尔),按照化合物44所述的合成步骤获得目标化合物46。快速柱色谱(石油醚∶乙酸乙酯=12∶1)得到化合物46(136毫克,68%,白色固体)。
[0221]
(44)化合物47的制备:
[0222]
化合物47的结构如下:
[0223]
其制备过程如下:
[0224]
使用40(200毫克,0.602毫摩尔),按照化合物45所述的合成步骤获得目标化合物47。快速柱色谱(石油醚∶乙酸乙酯=18∶1)得到化合物47(163毫克,82%,白色固体)。
[0225]
效果验证例:
[0226]
一、二萜衍生物的体外癌细胞抑制作用
[0227]
采用四甲基偶氮唑蓝(mtt)比色法检测五环三萜衍生物对癌细胞的抑制作用。
[0228]
mtt比色法实验步骤:将处于对数生长期的癌细胞按照每毫升5
×
104细胞数的密度接种至96孔细胞培养板中,调零孔为不含细胞的正常培养基。12小时后更换含不同浓度梯度异土五环三萜衍生物的培养基,调零孔更换正常培养基,每个浓度梯度设置5个复孔,置于37℃,5%co2培养箱中培养。24小时后显微镜下观察细胞状态及生长变化。48小时后每孔加入四甲基偶氮唑蓝(凯基生物,5mg/ml)溶液,继续在37℃,5%co2培养箱中培养,4小时后吸走培养基,每孔加入dmso 100μl,使用酶标仪在570纳米波长下测量吸光度值,使用graphpad软件进行数据统计分析,计算半数有效浓度(ic
50
,单位:μm)。抑制效果见下表1所示。
[0229]
表1二萜衍生物对不同癌细胞系增殖的影响
[0230]
[0231][0232]
其中,4t1,a375,b16f10,hct
‑
116,ct
‑
26,kasumi
‑
1分别表示小鼠乳腺瘤细胞、人恶性黑色素瘤细胞、小鼠黑色素瘤细胞、人结直肠腺癌细胞、小鼠结肠癌细胞、人急性原粒细胞白血病细胞。
[0233]
综上所述,与天然的截短侧耳素相比,这些合成类似物的抗增殖作用明显增强(至少比截短侧耳素强50倍)。恶性黑色素瘤细胞b16f10对合成化合物的治疗作用最敏感。化合物15、17、22和38具有较好的抗癌活性,因此用于后续研究。
[0234]
二、二萜衍生物的抗菌活性检测
[0235]
为了验证上述合成的二萜衍生物是否仍具有抗菌活性,本试验确定了所选化合物对金黄色葡萄球菌、大肠杆菌和枯草芽孢杆菌的最低抑菌浓度(mic),其中截短侧耳素作为
阳性对照(表2)。
[0236]
表2抗菌活性
[0237][0238]
正如所预期的情况,截短侧耳素是一种强效的广谱抗生素,然而所选的类似物无抗菌活性(mic>1000μg/ml)。基于上述结果,对截短侧耳素的结构修饰可以显著提高其抗癌活性,并降低其抗菌活性。
[0239]
三、体内抗肿瘤作用
[0240]
试验对象:本试验选用动物品系为c57bl/6j小鼠,雌性,18~22g,spf级别。
[0241]
试验接种内容:b16f10细胞皮下荷瘤。
[0242]
方法为小鼠皮下荷瘤法,将培养好的肿瘤细胞逐一接种到小鼠背部腋下处。待种瘤部位观察到明显瘤体后重新分组,保证瘤体大小以及体重均匀分布于各组之中,模型对照组小鼠每天给予0.1ml生理盐水,化合物38的给药剂量为20mg/kg。均采用腹腔注射给药治疗。每天记录一次小鼠体重和肿瘤大小(v=ab2/2,a为肿瘤长径,b为肿瘤短径),给药两周之后处死小鼠并采集肿瘤。结果如图1所示,其中图1(a)为b16f10异种移植瘤体积曲线,图1(b)为定量分析肿瘤重量的情况图,图1(c)为小鼠体重定量分析情况图,图1(d)为从小鼠模型中摘除的肿瘤图像。
[0243]
结果表明,化合物38能明显抑制肿瘤生长,且对小鼠体重的影响微乎其微。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1