一种双联苄地钱素S衍生物及其制备方法和应用
一种双联苄地钱素s衍生物及其制备方法和应用
技术领域
1.本技术涉及医药技术领域,具体涉及一种双联苄地钱素s衍生物及其制备方法和应用。
背景技术:
2.公开该背景技术部分的信息仅仅旨在增加对本技术的总体背景的理解,而不必然被视为承认或以任何形式暗示该信息构成已经成为本领域一般技术人员所公知的现有技术。
3.炎症是机体对于刺激的一种防御反应。任何能够引起组织损伤的因素,如生物性因素、物理性因素和化学性因素都能诱发炎症。根据持续时间不同,炎症可以分为急性炎症和慢性炎症。急性炎症一般只表现为血管性反应,是机体对来自体内或体外刺激的正常的防御反应,对机体是有益的。但若急性炎症反应不能实现排除刺激的影响,急性炎症反应相关的致炎因子将会持续存在并持续产生,如白细胞介素、趋化因子、细胞黏附分子以及炎症相关酶等,导致慢性炎症的发生。在慢性炎症中,致炎因子的持续存在造成组织损伤,而组织损伤又将进一步引发炎症,形成恶性循环,从而诱发多种疾病,对机体极为有害。
4.风湿性关节炎是一种常见的急性或慢性结缔组织炎症,可反复发作并累及心脏。临床上以关节和肌肉游走性酸楚和疼痛为特征,属变态反应性疾病。据统计,我国50岁以上人群中半数患风湿性关节炎,65岁以上人群中约85%患风湿性关节炎,且人数和比例仍在不断增加,发病形式相当严峻。风湿性关节炎给患者的行动带来了不便,严重影响了患者的生活质量。风湿性关节炎的治疗方法有多种,其中,抗炎药物在其治疗过程中具有重要作用。因此,发现和研制抗炎药物对于治疗风湿性关节炎具有重大意义。
5.此外,炎症还与肿瘤的发生具有密切联系,据估计约有15%的癌症是由慢性炎症引起的,炎症在肿瘤的发生和发展过程中具有重要作用。因此,抑制炎症是预防和治疗肿瘤的一种有效措施。
6.双联苄是苔藓植物中所特有的一类天然多酚化合物,具有抗真菌、抗微生物、抗氧化、细胞毒、昆虫拒食、植物生长调节和抗血小板凝集等广泛而又显著的生物学活性,是发现和研制新药的重要来源。
技术实现要素:
7.发明人在石地钱(reboulia hemisphaerica)中首次分离得到的具有全新结构的环状双联苄类化合物,呈无色块状结晶,分子式为c
29
h
26
o5,分子量为454,易溶于氯仿、乙酸乙酯等有机溶剂中,其结构式如下:
[0008][0009]
发明人将其命名为地钱素s(marchantin s),并对其进行了生物学活性研究,结果发现,研究发现地钱素s具有良好的抗炎和抗肿瘤活性,能够降低肿瘤细胞的促炎因子,诱导肿瘤细胞自噬性凋亡和衰老,可能成为一种新型的抗肿瘤药物或抗炎药物,具有相当广阔的研究前景和发展潜力,本技术进一步提供了地钱素s的全合成方法,并针对其进行了构效研究,提供了一系列具有抗炎和抗肿瘤活性的化合物,对于发现高效的抗炎和抗肿瘤药物具有十分重要的意义。
[0010]
具体地,本发明的技术方案如下所述:
[0011]
在本发明的第一方面,本发明提供了一种双联苄地钱素s衍生物及其药学上可接受的盐,所述衍生物具有式i所示结构:
[0012][0013]
r1、r2各自独立地选自氢、甲基、
‑
(ch2)
n
r9、
‑
(c=o)r9;
[0014]
r3为h或
‑
(ch2)
n
r9;
[0015]
r5、r6、r7、r8各自独立地选自氢、羟基、甲基、卤素、硝基和氨基;
[0016]
r9选自氨基和5
‑
6元含氮环;所述5
‑
6元含氮环可以任意含有或不含有另外的杂原子氧原子或氮原子;所述氨基和5
‑
6元含氮环可以是未被取代的或者是被选自c1‑5烷基、c1‑5卤代烷基、羟基、苯基、被c1‑5烷氧基或卤素取代的苯基、r
10
s(=o)2‑
中的至少一个取代基所取代的;
[0017]
r
10
选自c1‑5烷基、5
‑
6元含氮环;
[0018]
n为1或2;
[0019]
其中,r1、r2不同时为甲基。
[0020]
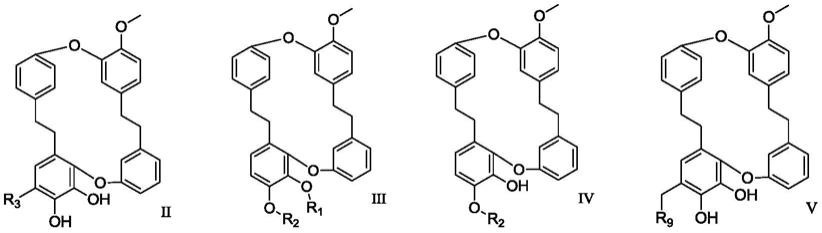
进一步地,在本发明的实施方式中,所述衍生物具有下述式ii、式iii、式iv或式v所示结构:
[0021][0022]
其中,式iii结构中,r1选自氢、
‑
(ch2)
n
r9、
‑
(c=o)r9;
[0023]
式iii和式iv结构中,r2选自
‑
(ch2)
n
r9、
‑
(c=o)r9;
[0024]
式ii结构中,r3为
‑
(ch2)
n
r9;
[0025]
r9、n同上文中所定义。
[0026]
在本发明的实施方式中,发明人发现当化合物符合式ii、式iii、式iv或式v所示结构时,具有抗肿瘤和/或抗炎活性。
[0027]
进一步地,在本发明的实施方式中,r9选自氨基和5
‑
6元含氮环,所述5
‑
6元含氮环选自吗啉基、哌嗪基、哌啶基和吡咯烷基;所述氨基和5
‑
6元含氮环可以是未被取代的或者是被选自甲基、乙基、c1‑2卤代烷基、羟基、苯基、甲氧基苯基、卤代苯基、甲基磺酰基、吗啉磺酰基中的至少一个取代基所取代的。
[0028]
尤其是,在本发明的实施方式中,发明人发现当本发明的化合物具有以下结构(式vi、vii或viii)时,往往具有特别优异的抗肿瘤和/或抗炎活性:
[0029][0030]
其中,环a为含氮脂肪环,环a选自吗啉基、哌嗪基、哌啶基和吡咯烷基;
[0031]
x选自c、n或o;
[0032]
r
11
选自氢、甲基、羟基、苯基、甲氧基苯基、卤代苯基、甲基磺酰基、吗啉磺酰基;
[0033]
r
12
、r
13
各自独立地选自c1‑5烷基或c1‑5卤代烷基,优选选自甲基、以及卤代乙基。
[0034]
尤其是,当环a为吗啉基、哌嗪基和哌啶基时,即所述化合物具有式vii所示结构(x选自c、n或o)时,往往具有特别优异的抗炎活性。
[0035]
尤其是,当r
12
和r
13
不相同时,化合物具有更优的活性。
[0036]
在本发明的一些实施方式中,式vi结构中,环a选自吗啉基、哌啶基和吡咯烷基时,r
11
为氢;环a为哌嗪基时,r
11
选自甲基、羟基、苯基、甲氧基苯基、卤代苯基、甲基磺酰基、吗啉磺酰基;优选地,r
11
取代位置为哌嗪基的n端。
[0037]
进一步地,上述式iii化合物中,r1和r2相同,且选自
‑
(ch2)
n
r9或
‑
(c=o)r9,所述化合物符合式ix或式x所示结构:
[0038][0039]
其中,r9选自吗啉基、哌啶基和吡咯烷基。
[0040]
进一步地,上述式iv化合物中,r2选自
‑
(ch2)
n
r9、
‑
(c=o)r9,r9选自吗啉基、哌嗪基、哌啶基和吡咯烷基。
[0041]
在本发明的实施方式中,r9为5
‑
6元含氮环时,含氮环以其氮端作为连接位点与亚甲基或羰基相连,在本发明通式结构下具有该连接特性的化合物具有显著更优的活性。
[0042]
进一步地,作为示例,本发明提供了一系列化合物,所述化合物如下所示:
[0043][0044]
在本发明的实施方式中,术语“盐”是指上述化合物或其立体异构体,与无机和/或
有机酸和碱形成的酸式和/或碱式盐,也包括两性离子盐(内盐),还包括季铵盐,例如烷基铵盐。这些盐可以是在化合物的最后分离和纯化中直接得到。也可以是通过将上述化合物,或其立体异构体,与一定数量的酸或碱适当(例如等当量)进行混合而得到。这些盐可能在溶液中形成沉淀而以过滤方法收集,或在溶剂蒸发后回收而得到,或在水介质中反应后冷冻干燥制得。比如,在一些实施方式中,本发明中所述盐可以是化合物的钾盐(koh,k2co3)、钠盐(naoh,na2co3)、钙盐(cacl2,ca(oac)2)和镁盐(mgcl2)等;或者所述化合物药学上可接受的盐为所述化合物与无机酸(盐酸、硫酸、氢溴酸)或有机酸(甲磺酸、甲苯磺酸、三氟乙酸)形成的盐。
[0045]
在本发明的第二方面,本发明提供了一种制备上述第一方面中所述的双联苄地钱素s衍生物及其药学上可接受的盐的方法,其包括按照以下反应路线进行:
[0046]
反应路线1:
[0047][0048]
其中,r1、r2、r3、r5、r6、r7、r8同上文中所定义。
[0049]
进一步地,所述方法还包括以化合物18为起始化合物行下述反应路线2或3:
[0050]
反应路线2:
[0051]
反应路线3:
[0052]
其中,r1、r2、r3、r5、r6、r7、r8、r9、r
11
、r
12
、r
13
同上文中所定义。
[0053]
在本发明的一些实施方式中,r1、r2选自甲基和氢原子,r5、r6、r7和r8各自独立地选自氢原子、羟基、烷氧基时,反应路线1涉及以下步骤:
[0054]
步骤a:将化合物1和化合物2以及氧化亚铜和碳酸钾,在吡啶中搅拌,加热,反应结束后,过滤、蒸除溶剂、硅胶柱层析纯化,获得化合物3;
[0055]
步骤b:冰水浴条件下,向化合物3的乙醇或四氢呋喃溶液中慢慢加入硼氢化钠,室温下磁力搅拌,反应结束后,蒸除溶剂,硅胶柱层析纯化,获得化合物4;
[0056]
步骤c:将化合物4溶于乙腈中,加入三苯基膦氢溴酸盐,搅拌,加热回流,反应结束后,蒸除溶剂,得到化合物5;
[0057]
步骤d:将化合物6和化合物7溶解于dmf溶液中,加入碳酸钾,加热,搅拌,反应结束后,蒸除溶剂,经硅胶柱层析纯化,得到化合物8;
[0058]
步骤e:将化合物5和化合物8溶解于二氯甲烷溶液中,加入碳酸钾和少量18
‑
冠
‑
6,加热回流,搅拌,反应结束后,蒸除溶剂,经硅胶柱层析纯化,得到化合物9;
[0059]
步骤f:将化合物9溶解于乙酸乙酯或甲醇溶液中,加入钯碳,氢气条件下,搅拌,反应结束后,蒸除溶剂,经硅胶柱层析纯化,得到化合物10;
[0060]
步骤g:将化合物10和亚硝酸异戊酯溶解于乙醚或四氢呋喃溶液中,加热回流,搅拌,反应结束后,蒸除溶剂,经硅胶柱层析纯化,得到化合物11;
[0061]
步骤h:将化合物11慢慢滴加至含有四氢铝锂的乙醚或四氢呋喃溶液中,加热回流,搅拌,反应完成后,慢慢加入氯化铵饱和溶液,二氯甲烷萃取,有机相用硫酸钠干燥,过滤,蒸除溶剂,经硅胶柱层析纯化,得到化合物12;
[0062]
步骤i:将化合物12溶解于无机酸的乙醇或四氢呋喃溶液中,搅拌,反应结束后,蒸除溶剂,经硅胶柱层析纯化,得到化合物13;
[0063]
步骤j:将化合物13和碳酸钾、溴化苄溶解于乙腈或dmf溶液中,搅拌,加热,反应结束后,抽滤,蒸除溶剂,经硅胶柱层析纯化,得到化合物14;
[0064]
步骤k:将化合物14和氯化亚砜、三乙胺溶解于二氯甲烷溶液中,搅拌,反应结束后,蒸除溶剂,经硅胶柱层析纯化,得到如化合物15;
[0065]
步骤l:将化合物15溶于乙腈中,加入三苯基膦,搅拌,加热,反应结束后,蒸除溶
剂,得到化合物16;
[0066]
m:将化合物16的二氯甲烷溶液慢慢滴加至甲醇钠或叔丁醇钠的二氯甲烷溶液中,搅拌,反应结束后,蒸除溶剂,经硅胶柱层析纯化,得到化合物17;
[0067]
步骤n:将化合物17溶解于乙酸乙酯或甲醇溶液中,加入钯碳,氢气条件下,搅拌,反应结束后,抽滤,蒸除溶剂,经硅胶柱层析纯化,得到化合物18。
[0068]
进一步地,在反应路线1结束后,以反应路线1的产物化合物18为起始物进行反应路线2或3,其中,反应路线2的步骤包括:将化合物18和碳酸钾、碘甲烷溶解于丙酮溶液中,搅拌,加热,反应结束后,抽滤,蒸除溶剂,经硅胶柱层析纯化,得到化合物19;低温条件下,将三溴化硼滴加至化合物19的二氯甲烷溶液中,搅拌,反应结束后,加入碳酸氢钠水溶液,并用二氯甲烷萃取,合并有机相,硫酸钠干燥,过滤,蒸除溶剂,经硅胶柱层析纯化,得到化合物21;
[0069]
其中,反应路线3的步骤包括:将化合物18和具有不同取代基的仲胺化合物r9h或r9cl以及甲醛溶解到二氧六环溶液中,搅拌,加热,反应结束后,蒸除溶剂、硅胶柱层析纯化,获得化合物22或27。其中,r1、r2为氢,r3同上文中所定义。
[0070]
或者,在本发明的有一些实施方式中,r1、r2选自氢、
‑
(ch2)
n
r9、
‑
(c=o)r9,r5、r6、r7和r8各自独立地选自氢原子、羟基、烷氧基时,以化合物18为起始产物进行反应路线4:
[0071][0072]
其中,r9同上文中所定义;反应路线4涉及以下步骤:将化合物18和具有不同取代氨基的氯代烷盐酸盐以及碳酸钾溶解到丙酮溶液中,搅拌,加热,反应结束后,过滤、蒸除溶剂、硅胶柱层析纯化,可以分别获得一系列化合物21f~k;
[0073]
将化合物18和吗啉甲酰氯、三乙胺、dmap溶解至二氯甲烷溶液中,搅拌,加热,反应结束后,蒸除溶剂,经硅胶柱层析纯化,可以分别获得化合物21d、21e。
[0074]
在本发明的一些实施方式中,本发明提供了地钱素s的全合成方法,其包括按照以下反应路线进行合成:
[0075][0076]
本领域技术人员能够基于本发明提供的反应路线通过有限次摸索制备得到本发明的化合物。需知的,在反应路线的公开下,选取合适的溶剂以及获取进一步提高的收率和纯净度都是本领域的合理的常规的追求,这种追求可在掌握有本领域常规技术知识的前提下可通过有限次实验尝试实现。
[0077]
在本发明的第三方面,本发明提供了一种药物组合物,其包含上述第一方面中所述的双联苄地钱素s衍生物及其药学上可接受的盐。
[0078]
以及,本发明提供了一种药物制剂,其包含上述第一方面中所述的双联苄地钱素s衍生物及其药学上可接受的盐和至少一种药学上可接受的辅料或药物载体。
[0079]
本发明所述药物组合物或药物制剂可以为固体口服制剂、液体口服制剂或注射剂,比如可以为片剂、分散片、肠溶片、咀嚼片、口崩片、胶囊、糖衣剂、颗粒剂、干粉剂、口服溶液剂、注射用小水针、注射用冻干粉针、大输液或小输液。
[0080]“药学上可接受的辅料”是指药物组合物中除有效成分之外的成分,其对受试者无毒。比如赋形剂,药学上可接受的赋形剂包括但不限于缓冲剂、稳定剂或防腐剂等。本发明所述的药学上可接受的载体和辅料通常为本领域人员所熟知的,或者可由本领域技术人员根据实际情况能够确定的。
[0081]“药物载体”是指药学上可接受的溶剂,悬浮剂或载体,用于将化合物递送至动物或人体内。载体可以是液体或固体,并按照计划的给药方式进行选择。蛋白和脂质体也是药物载体。载体在药物组合物中的含量可以是1wt%
‑
98wt%,通常大约占到80wt%。
[0082]
本发明的一些实施方式中包括生产药物组合物或药物制剂的方法,所述方法包括将根据本发明公开的至少一种化合物与药用辅料或载体混合。通过任意合适的方法制备制
剂,通常通过以所需比例均匀混合活性化合物与液体和/或微细粉碎的固体辅料制备,然后如果需要,使所得混合物形成所需的形状。可使用本领域技术人员公知的技术将本发明的化合物配制成药物组合物或制剂。比如可根据由沈阳药科大学主编的现代药物制剂丛书进行药物制剂的制备。除本文提到以外,合适的药用辅料是本领域已知的,例如参见2005年版的药用辅料手册(原著第四版),作者(英)r.c.罗(raymondcrowe)(美)p.j.舍斯基(pauljsheskey)。
[0083]
在本发明的第四方面,本发明提供了上述第一方面中所述的双联苄地钱素s衍生物及其药学上可接受的盐、或者上述第三方面中药物组合物或者药物制剂在制备抗肿瘤药物和/或抗炎中的应用。
[0084]
在本发明的一些实施方式中,本发明所述肿瘤包括但不限于肝癌、非小细胞肺癌、乳腺癌、前列腺癌、支气管上皮样细胞类癌症。
[0085]
在本发明的一些实施方式中,本发明所述抗炎表现为对促炎因子白介素
‑
1β(il1β)转录水平(基因表达水平)和/或促炎因子白介素
‑
6(il6)转录水平(基因表达水平)的抑制作用。
[0086]
在本发明的第五方面,本发明提供了上述第一方面中所述的双联苄地钱素s衍生物及其药学上可接受的盐、或者上述第三方面中药物组合物或者药物制剂在制备用于治疗和/或缓解风湿性关节炎的药物或者用于治疗和/或缓解由炎症引发的肿瘤的药物中的应用。
[0087]
在本发明的第六方面,本发明提供了化合物20或其药学上可接受的盐或包含该化合物或其盐的药物组合物或药物制剂在制备抗炎药物中的应用,或者在制备用于治疗和/或缓解风湿性关节炎的药物和/或缓解由炎症引发的肿瘤的药物中的应用;
[0088][0089]
在本发明的第六方面,本发明提供了一种治疗疾病的方法,其包括向受试者施用有效剂量的本发明上述第一方面中所述的双联苄地钱素s衍生物及其药学上可接受的盐、或者上述第三方面中药物组合物或者药物制剂或化合物20以及包含化合物20的组合物或药物制剂。
[0090]
所述疾病包括肝癌、非小细胞肺癌、乳腺癌、前列腺癌、支气管上皮样细胞类癌症以及风湿性关节炎和由炎症引发的肿瘤。
[0091]
如本文所用,“治疗”是指试图改变所治疗个体的自然进程的临床干预,并且可以是为了预防或在临床病理学的进程中进行。治疗的期望效果包括但不限于预防疾病的发生或复发、减轻症状、削弱疾病的任何直接或间接病理学后果、预防转移、降低疾病进展的速率、改善或减轻疾病状态,以及缓解或改善预后。
[0092]“受试者”是指已经是治疗、观察或实验的对象的动物,优选指哺乳动物,最优选指
人。
[0093]“治疗有效量”是指包括本发明化合物在内的活性化合物或药剂的量,该量可引起研究者、兽医、医生或其他医疗人员所追求的组织系统、动物或人的生物学或医学响应,这包括减轻或部分减轻受治疗的疾病、综合征、病症或障碍的症状。必须认识到,本发明化合物的最佳给药剂量和间隔是由化合物性质和诸如给药的形式、路径和部位以及所治疗的特定哺乳动物等外部条件决定的,而这一最佳给药剂量可用常规的技术确定。同时也必须认识到,最佳的疗程,即同时化合物在额定的时间内每日的剂量,可用本领域内公知的方法确定。
[0094]
相较于现有技术,本发明的优势在于:本发明首次在石地钱(reboulia hemisphaerica)中分离得到的具有全新结构的环状双联苄类化合物地钱素s(marchantin s),经实验发现地钱素s具有优异的抗肿瘤活性和抗炎活性,在此基础上本发明设计合成了一些列具有地钱素s样结构并且具有抗肿瘤和/或抗炎活性的的地钱素s衍生物。
具体实施方式
[0095]
下面结合具体实施例,进一步阐述本技术。应理解,这些实施例仅用于说明本技术而不用于限制本技术的范围。下列实施例中未注明具体条件的实验方法,通常按照常规条件或按照制造厂商所建议的条件。
[0096]
除非另行定义,文中所使用的所有专业与科学用语与本领域熟练人员所熟悉的意义相同。本技术所使用的试剂或原料均可通过常规途径购买获得,如无特殊说明,本技术所使用的试剂或原料均按照本领域常规方式使用或者按照产品说明书使用。此外,任何与所记载内容相似或均等的方法及材料皆可应用于本技术方法中。文中所述的较佳实施方法与材料仅作示范之用。
[0097]
实施例1地钱素s的全合成
[0098]
合成路线如下所示:
[0099][0100]
步骤b:化合物3’的制备
[0101]
将化合物2’(80g)、对溴苯甲醛(70g)、碳酸钾(100g)和氧化铜(10g)的混合物在吡啶(250ml)中回流搅拌,反应结束后,蒸除吡啶,二氯甲烷萃取残余物。真空浓缩溶液,通过硅胶柱层析纯化残余物,用etoac
‑
pe溶液洗脱,得到无色油状产物。1h nmr(400mhz,cdcl3)δ9.88(s,1h),7.79(d,j=8.7hz,2h),7.35(dd,j=8.5,1.8hz,1h),7.25(d,j=1.9hz,1h),7.00(d,j=8.5hz,1h),6.97(d,j=8.6hz,2h),5.45(s,1h),4.23(dd,j=10.9,4.9hz,2h),3.95(td,j=12.3,2.2hz,2h),3.77(s,3h),2.18(qt,j=12.5,5.0hz,1h),1.42(d,j=13.5hz,1h).
[0102]
步骤c:化合物4’的制备
[0103]
低温条件下,将硼氢化钠(35g)添加到化合物3’(120g)的无水乙醇溶液(1200ml)中。将所得混合物在室温下搅拌1
‑
3h。0℃下缓慢添加水淬灭反应,并蒸除乙醇。所得混合物用etoac萃取,并用硫酸钠干燥有机相。过滤除掉不溶性物质,真空浓缩溶液,通过硅胶柱层析纯化残余物,用etoac
‑
pe溶液洗脱,得到白色油状产物。1h nmr(400mhz,cdcl3)δ7.26
–
7.22(m,3h),7.10(d,j=2.0hz,1h),6.97(d,j=8.4hz,1h),6.90(d,j=8.6hz,2h),5.38(s,1h),4.57(s,2h),4.19(dd,j=10.8,5.0hz,2h),3.95
–
3.89(m,2h),3.80(s,3h),2.13(ddd,j=12.5,7.9,5.0hz,1h),1.42
–
1.35(m,1h).
[0104]
步骤d:化合物5’的制备
[0105]
将化合物4’(110g)和三苯基膦氢溴酸盐(150g)溶解于mecn(1100ml)中,回流搅拌1
‑
3h,真空浓缩溶液,通过硅胶柱层析纯化残余物,用meoh
‑
dcm溶液洗脱,得到白色固体,mp 131
‑
132℃。1h nmr(400mhz,cdcl3)δ7.72
–
7.64(m,10h),7.58(dd,j=7.7,3.3hz,5h),7.20
–
7.15(m,1h),7.01
–
6.98(m,1h),6.94(s,1h),6.92(d,j=2.2hz,1h),6.89(d,j=8.5hz,1h),6.63(d,j=8.3hz,2h),5.37(s,1h),5.36(s,1h),5.33(s,1h),4.14(dd,j=11.3,4.4hz,2h),3.93
–
3.85(m,2h),3.73(s,3h),2.15
–
2.05(m,1h),1.40
–
1.33(m,1h).
[0106]
步骤e:化合物7’的制备
[0107]
将n,n
‑
二异丙基乙胺(122g)滴加到2,3,4
‑
三羟基苯甲醛(58.00g)的dcm溶液(600ml)中。将所得混合物在室温下搅拌0.5
‑
2h。在0℃下向混合物中缓慢添加氯甲基乙醚(85g),在室温下将所得混合物搅拌12h。用饱和食盐水洗涤反应混合物,用无水硫酸钠干燥有机相。过滤除掉不溶性物质,真空浓缩溶液,通过硅胶柱层析纯化残余物,用etoac
‑
pe溶液洗脱,得到黄色油状产物。1h nmr(400mhz,cdcl3)δ11.23(s,1h),9.71(s,1h),7.23(d,j=8.8hz,1h),6.81(d,j=8.8hz,1h),5.29(s,2h),5.18(s,2h),3.88(q,j=7.1hz,2h),3.71(q,j=7.0hz,2h),1.20
–
1.18(m,3h),1.18
–
1.15(m,3h).
[0108]
步骤f:化合物8’的制备
[0109]
将5
‑
氟
‑2‑
硝基苯甲酸甲酯(52g,)添加到化合物7(67.00g)和叔丁醇钾(42g)的dmf(500ml)溶液中。将所得混合物在80℃下搅拌2
‑
8h。加入etoac(2000ml)后用饱和食盐水(300ml
×
3)洗涤反应混合物,无水硫酸钠干燥有机相。过滤除掉不溶性物质,真空浓缩溶液,通过硅胶柱层析纯化残余物,用etoac
‑
pe溶液洗脱,得到黄色油状产物。1h nmr(400mhz,cdcl3)δ10.01(s,1h),8.00(d,j=9.0hz,1h),7.72(d,j=8.8hz,1h),7.28(d,j=8.9hz,1h),7.07(d,j=2.6hz,1h),7.01(dd,j=9.0,2.7hz,1h),5.36(s,2h),5.10(s,2h),3.90(s,3h),3.77(q,j=7.0hz,2h),3.67(q,j=7.1hz,2h),1.25(t,j=7.0hz,3h),1.16(t,j=7.1hz,3h).
[0110]
步骤g:化合物9’的制备
[0111]
将碳酸钾(22g)和微量18
‑
冠醚
‑
6添加到化合物5’(55g)和化合物8’(35g)的无水dcm溶液(1000ml)中,将所得混合物回流搅拌5
‑
12h。过滤除掉不溶性物质,真空浓缩溶液,通过硅胶柱层析纯化残余物,用etoac
‑
pe溶液洗脱,得到顺反异构的黄色油状产物。
[0112]
步骤h:化合物10’的制备
[0113]
将25%的pd/c(400mg)和三乙胺(100μl)添加到化合物9’(1.5g)的etoac溶液(60ml)中。将悬浮液通入氢气,在室温下搅拌2
‑
8h。过滤除掉不溶性物质,真空浓缩溶液,通过硅胶柱层析纯化残余物,用etoac
‑
pe溶液洗脱,得到黄色油状物。1h nmr(400mhz,cdcl3)δ7.31(d,j=3.0hz,1h),7.24(dd,j=8.4,2.0hz,1h),7.08(d,j=2.0hz,1h),7.00(d,j=5.4hz,1h),6.98(d,j=2.3hz,2h),6.96(d,j=5.3hz,1h),6.88(d,j=8.6hz,1h),6.84(dd,j=8.9,3.0hz,1h),6.81(d,j=8.5hz,2h),6.58(d,j=8.9hz,1h),5.45(s,2h),5.39(s,1h),5.24(s,2h),5.08(s,2h),4.20(dd,j=10.8,4.9hz,2h),3.93(dd,j=12.3,2.3hz,2h),3.82(s,3h),3.80(s,3h),3.79
–
3.74(m,2h),3.75
–
3.69(m,2h),2.73(s,4h),2.23
–
2.09(m,1h),1.42
–
1.36(m,1h),1.24(t,j=7.1hz,3h),1.17(t,j=7.1hz,3h).
[0114]
步骤i:化合物11’的制备
[0115]
在60℃下将化合物10’(11g)的无水thf溶液(20ml)逐滴添加到亚硝酸异戊酯(5g)的无水thf溶液(10ml)中。将所得混合物回流搅拌4
‑
9h。真空浓缩溶液,通过硅胶柱层析纯化残余物,用etoac
‑
pe溶液洗脱,得到黄色油状产物。1h nmr(400mhz,cdcl3)δ7.67(d,j=7.7hz,1h),7.53
–
7.48(m,1h),7.32(t,j=8.0hz,1h),7.24(dd,j=8.4,1.9hz,1h),7.07
(d,j=2.0hz,1h),7.03(d,j=8.6hz,1h),6.99(dd,j=8.2,2.0hz,1h),6.97(d,j=3.9hz,1h),6.95(d,j=4.0hz,2h),6.91(d,j=8.6hz,1h),6.80(d,j=8.5hz,2h),5.38(s,1h),5.25(s,2h),5.07(s,2h),4.20(dd,j=10.8,4.9hz,2h),3.93(dd,j=12.3,2.3hz,2h),3.87(s,3h),3.81(s,3h),3.76(q,j=7.1hz,2h),3.68(q,j=7.1hz,2h),2.73(d,j=6.0hz,4h),2.23
–
2.09(m,1h),1.42
–
1.36(m,1h),1.24(t,j=7.0hz,3h),1.15(t,j=7.1hz,3h).
[0116]
步骤j:化合物12’的制备
[0117]
在室温下将化合物11’(7g)的无水thf溶液(20ml)逐滴添加到氢化铝锂(1g)的无水thf悬浮液(20ml)中。将所得混合物回流搅拌1
‑
3h,然后冷却至0℃。缓慢加入水淬灭反应至胶体状态。用漏斗过滤并用etoac洗涤,真空浓缩溶液,通过硅胶柱层析纯化残余物,用etoac
‑
pe溶液洗脱,得到无色油状产物。1h nmr(400mhz,cdcl3)δ7.25(d,j=1.8hz,1h),7.23
–
7.21(m,1h),7.07(d,j=7.1hz,1h),7.02(d,j=8.5hz,1h),6.98(s,1h),6.96(s,2h),6.94(s,1h),6.91(d,j=8.7hz,1h),6.80(s,1h),6.78(s,2h),6.76(d,j=8.3hz,1h),5.38(s,1h),5.24(s,2h),5.08(s,2h),4.60(s,2h),4.20(dd,j=11.4,4.6hz,2h),3.92(t,j=11.9hz,2h),3.82(s,3h),3.76(q,j=7.1hz,2h),3.70(q,j=7.0hz,2h),2.72(s,4h),2.22
–
2.11(m,1h),1.39(d,j=13.5hz,1h),1.25(t,j=6.6hz,3h),1.15(t,j=7.1hz,3h)。
[0118]
步骤k:化合物13’的制备
[0119]
将化合物12’(3.5g)溶解于乙醇(80ml)和盐酸溶液(30ml)中,在室温下将所得混合物搅拌3
‑
6h。逐滴添加饱和碳酸氢钠溶液,在真空中蒸馏出乙醇。所得混合物用dcm萃取收集有机相,用无水硫酸钠干燥有机相。过滤除掉不溶性物质,真空浓缩溶液,通过硅胶柱层析纯化残余物,用etoac
‑
pe溶液洗脱,得到无色油状产物。1h nmr(400mhz,cdcl3)δ9.69(s,1h),7.55(dd,j=8.3,1.9hz,1h),7.31(d,j=1.9hz,1h),7.20(t,j=7.9hz,1h),7.08(d,j=8.3hz,1h),6.97(d,j=7.6hz,1h),6.94(d,j=8.5hz,2h),6.88(d,j=8.5hz,2h),6.80
–
6.74(m,2h),6.69
–
6.65(m,2h),6.22(s,1h),5.62(s,1h),4.58(s,2h),3.99(s,3h),2.76(t,j=7.2hz,2h),2.63(t,j=7.3hz,2h)。
[0120]
步骤l:化合物14’的制备
[0121]
将碳酸钾(2g)和溴化苄(2g)添加到化合物13’(1.5g)的乙腈溶液(30ml)中,将所得混合物回流搅拌1
‑
5h。过滤除掉不溶性物质,真空浓缩溶液,通过硅胶柱层析纯化残余物,用etoac
‑
pe溶液洗脱,得到无色油状产物。1h nmr(400mhz,cdcl3)δ9.79(s,1h),7.61(d,j=8.3hz,1h),7.42(d,j=7.3hz,2h),7.37(d,j=8.1hz,2h),7.34(d,j=4.3hz,1h),7.31(d,j=7.7hz,1h),7.23(d,j=8.2hz,1h),7.22
–
7.14(m,5h),7.08(d,j=8.3hz,1h),7.03(s,1h),7.01(s,1h),6.97(d,j=7.4hz,1h),6.88
–
6.83(m,5h),6.80(d,j=7.8hz,1h),5.11(s,2h),4.94(s,2h),4.59(d,j=4.8hz,2h),3.95(s,3h),2.80(s,4h)。
[0122]
步骤m:化合物15’的制备
[0123]
低温条件下,将氯化亚砜(250mg)和微量三乙胺添加到化合物14’(680mg)的dcm溶液(20ml)中,室温搅拌2
‑
8h,蒸除溶剂。将所得无色油状产物和三苯基膦(400mg)溶解于乙腈(20ml)中,回流搅拌10
‑
30h,真空浓缩溶液,通过硅胶柱层析,用meoh
‑
dcm溶液洗脱,得到白色固体,mp 104
‑
105℃。1h nmr(400mhz,cdcl3)δ9.76(s,1h),7.64
–
7.57(m,10h),7.47(dd,j=7.5,3.3hz,5h),7.42(d,j=7.3hz,3h),7.36(dd,j=14.2,6.5hz,4h),7.20(q,j=
6.2hz,3h),7.14(d,j=6.6hz,2h),7.09(d,j=8.4hz,1h),7.04(d,j=8.0hz,1h),6.98(d,j=8.3hz,2h),6.85(d,j=8.3hz,3h),6.80(dd,j=14.2,5.8hz,3h),6.32(s,1h),5.24(d,j=14.5hz,2h),5.08(s,2h),4.80(s,2h),3.93(s,3h),2.72
–
2.66(m,2h),2.61
–
2.55(m,2h)。
[0124]
步骤o:化合物16’的制备
[0125]
将化合物15’(1.3g)的无水dcm溶液(200ml)慢慢滴加到甲醇钠(1g)的无水dcm悬浮液(150ml)中,搅拌6
‑
24h。过滤、真空浓缩,通过硅胶柱层析纯化,用dcm
‑
pe溶液洗脱,得到顺反异构的白色固体。
[0126]
步骤p:化合物18’(地钱素s)的制备
[0127]
将pd/c(200mg)加入到化合物16’(800mg)的etoac溶液(30ml)中。将悬浮液通入氢气,在室温下搅拌1
‑
4h。过滤除掉不溶性物质,真空浓缩溶液,通过硅胶柱层析纯化残余物,用etoac
‑
pe溶液洗脱,得到白色固体,mp 83
‑
84℃。1h nmr(400mhz,cdcl3)δ6.98(t,j=7.8hz,1h),6.93(d,j=6.9hz,2h),6.91(s,1h),6.88(s,1h),6.85(d,j=6.5hz,1h),6.78(d,j=8.1hz,1h),6.65(s,1h),6.61(d,j=7.6hz,2h),6.55(d,j=8.2hz,1h),6.35(d,j=7.4hz,1h),5.51(s,1h),4.72(s,1h),3.90(s,3h),2.97(d,j=7.3hz,2h),2.93(d,j=7.5hz,2h),2.90
–
2.81(m,2h),2.82
–
2.73(m,2h)。
[0128]
实施例2化合物20和21a~21c的合成
[0129]
化合物20的制备:
[0130][0131]
低温条件下,将1m bbr3(300mg)的无水dcm溶液(1.2ml)缓慢滴入地钱素s(化合物18’)(70mg)的无水dcm溶液中,反应混合物搅拌1
‑
2h。逐滴添加饱和碳酸氢钠溶液淬灭反应,用饱和食盐水洗涤反应混合物,无水硫酸钠干燥有机相。过滤,真空浓缩溶液,通过硅胶柱层析纯化,用etoac
‑
pe溶液洗脱,得到白色固体,mp 79
‑
80℃。1h nmr(400mhz,cdcl3)δ6.99(d,j=7.9hz,1h),6.96(s,1h),6.93(d,j=7.4hz,2h),6.90
–
6.85(m,2h),6.74(dd,j=8.1,1.9hz,1h),6.64
–
6.62(m,1h),6.60(d,j=8.4hz,2h),6.55(dd,j=8.2,2.1hz,1h),6.38(d,j=7.6hz,1h),5.55(s,1h),5.54(d,j=1.9hz,1h),5.40(s,1h),4.84(s,1h),2.99(dd,j=6.7,3.7hz,2h),2.93(dd,j=6.7,3.8hz,2h),2.89
–
2.83(m,2h),2.81
–
2.75(m,2h)。
[0132]
化合物21a~21c的制备:
[0133][0134]
将碳酸钾(70mg)和碘甲烷(50mg)加入到地钱素s(80mg)的丙酮溶液(15ml)中,将所得混合物回流搅拌6h。过滤,真空浓缩溶液,通过硅胶柱层析纯化,用etoac
‑
pe溶液洗脱,得到化合物21a、21b、21c三个白色固体。
[0135]
化合物21a,mp 168
‑
169℃,1h nmr(400mhz,cdcl3)δ7.09(d,j=8.5hz,1h),6.94(d,j=5.9hz,1h),6.92(d,j=6.7hz,2h),6.88(d,j=8.5hz,1h),6.84(d,j=8.2hz,1h),6.77(dd,j=8.2,1.9hz,1h),6.64
–
6.62(m,1h),6.61(s,1h),6.59(s,1h),6.51(dd,j=8.2,2.0hz,1h),6.30(d,j=7.5hz,1h),5.67(s,1h),5.52(d,j=1.9hz,1h),3.90(s,3h),3.61(s,3h),2.98(s,4h),2.84(dd,j=8.3,3.3hz,2h),2.80
–
2.75(m,2h).
[0136]
化合物21b,mp 66
‑
67℃。1h nmr(600mhz,cdcl3)δ6.95(d,j=8.2hz,2h),6.92(d,j=5.6hz,2h),6.84(d,j=8.2hz,1h),6.82(d,j=8.5hz,1h),6.77(d,j=8.2hz,1h),6.66(s,1h),6.61(d,j=8.1hz,2h),6.55
–
6.51(m,1h),6.28(d,j=7.3hz,1h),5.50(d,j=8.8hz,1h),5.37(s,1h),3.91(d,j=9.7hz,6h),2.98(d,j=8.4hz,4h),2.86
–
2.81(m,2h),2.77(d,j=6.1hz,2h).
[0137]
化合物21c,mp 67
‑
68℃。1h nmr(400mhz,cdcl3)δ7.13(d,j=8.6hz,1h),6.91(d,j=7.4hz,2h),6.85(d,j=2.5hz,1h),6.83(d,j=3.0hz,1h),6.77(dd,j=8.2,1.9hz,1h),6.61(s,2h),6.59(s,1h),6.53(dd,j=8.2,2.0hz,1h),6.30(d,j=7.5hz,1h),5.57(d,j=1.8hz,1h),3.89(d,j=6.1hz,6h),3.59(s,3h),2.99(s,4h),2.83(dd,j=8.2,3.3hz,2h),2.76(dd,j=8.4,3.4hz,2h).
[0138]
实施例3化合物21d~21k的合成
[0139]
化合物21d和21e的制备:
[0140][0141]
将三乙胺(30mg)添加到地钱素s(50mg)的dcm溶液(10ml)中。将所得混合物在室温下搅拌5min。向混合物中加入4
‑
吗啉碳酰氯(25mg)和4
‑
二甲氨基吡啶(5mg),在室温下将所得混合物搅拌1
‑
3h。逐滴添加水淬灭反应,所得混合物用dcm萃取收集有机相,再用饱和食盐水洗涤,用无水硫酸钠干燥有机相。过滤除掉不溶性物质,真空浓缩溶液,通过硅胶柱层
析纯化残余物,用etoac
‑
pe溶液洗脱,得到化合物21d和21e两个白色固体。
[0142]
化合物21d,mp 100
‑
101℃。1h nmr(600mhz,cdcl3)δ7.24(d,j=8.6hz,1h),6.98(s,1h),6.93(s,1h),6.91(d,j=6.6hz,2h),6.83(d,j=8.2hz,1h),6.78
–
6.75(m,1h),6.57(d,j=7.9hz,2h),6.52(d,j=6.9hz,2h),6.27(d,j=7.4hz,1h),5.46
–
5.44(m,1h),3.89(s,3h),3.48
–
3.44(m,2h),3.32
–
3.28(m,2h),3.12
–
3.08(m,2h),2.99(s,4h),2.88
–
2.80(m,4h),2.71(s,2h).
[0143]
化合物21e,mp 172
‑
173℃。1h nmr(400mhz,cdcl3)δ7.33(d,j=8.6hz,1h),7.14(d,j=8.6hz,1h),6.95(d,j=8.3hz,2h),6.91(t,j=7.9hz,1h),6.83(d,j=8.2hz,1h),6.76(dd,j=8.2,1.8hz,1h),6.59(d,j=8.0hz,3h),6.53(dd,j=8.2,2.2hz,1h),6.26(d,j=7.4hz,1h),5.47(d,j=1.7hz,1h),3.89(s,3h),3.74
–
3.62(m,4h),3.62
–
3.50(m,4h),3.46(s,2h),3.29(s,2h),3.14
–
2.97(m,6h),2.89
–
2.79(m,4h),2.72(d,j=5.5hz,2h).
[0144]
化合物21f和21g的制备:
[0145][0146]
将碳酸钾(50mg)和n
‑
(2
‑
氯乙基)哌啶盐酸盐(30mg)添加到地钱素s(50mg,)的丙酮溶液(10ml)中,将所得混合物回流搅拌6h。过滤除掉不溶性物质,真空浓缩溶液,通过硅胶柱层析纯化残余物,用meoh
‑
dcm溶液洗脱,得到化合物21f和21g两个白色固体。
[0147]
化合物21f,mp 163
‑
164℃。1h nmr(600mhz,cdcl3)δ7.07(d,j=8.6hz,1h),6.95(d,j=8.3hz,2h),6.90(t,j=7.9hz,1h),6.86(d,j=8.6hz,1h),6.84(d,j=8.4hz,1h),6.77(dd,j=8.2,1.8hz,1h),6.65(s,1h),6.62(d,j=8.2hz,2h),6.48(dd,j=8.2,2.1hz,1h),6.24(d,j=7.5hz,1h),5.50(d,j=1.8hz,1h),3.90(s,3h),3.74
–
3.70(m,2h),2.99
–
2.95(m,2h),2.94
–
2.89(m,2h),2.84
–
2.80(m,2h),2.78
–
2.73(m,2h),2.58
–
2.31(m,6h),1.67(q,j=5.5hz,4h).
[0148]
化合物21g,mp 119
‑
120℃。1h nmr(600mhz,cdcl3)δ7.15(d,j=8.5hz,1h),6.95(d,j=7.6hz,2h),6.89(d,j=7.7hz,1h),6.86(d,j=7.9hz,1h),6.83(d,j=8.2hz,1h),6.76(d,j=8.2hz,1h),6.62(d,j=7.5hz,2h),6.60(s,1h),6.45(d,j=7.9hz,1h),6.24(d,j=7.2hz,1h),5.45(s,1h),4.31(s,4h),3.90(s,3h),3.35(s,4h),3.02
–
2.80(m,8h),2.79
–
2.36(m,8h),1.62(d,j=97.1hz,12h).
[0149]
化合物21h和21i的制备:
[0150][0151]
参考化合物21f和21g的合成方法。以地钱素s和n
‑
(2
‑
氯乙基)吗啉盐酸盐为原料,得到化合物21h和21i两个白色固体。
[0152]
化合物21h,mp 78
‑
79℃。1h nmr(400mhz,cdcl3)δ7.09(d,j=8.5hz,1h),6.97
–
6.92(m,2h),6.89(d,j=7.9hz,1h),6.84(dd,j=8.4,3.1hz,2h),6.76(dd,j=8.2,1.9hz,1h),6.62(d,j=8.4hz,3h),6.51
–
6.44(m,1h),6.25(d,j=7.5hz,1h),5.50(s,1h),3.90(s,3h),3.78(s,6h),3.00
–
2.95(m,2h),2.94
–
2.89(m,2h),2.82(dd,j=7.9,3.3hz,2h),2.78
–
2.71(m,2h),2.61
–
2.48(m,4h),2.48
–
2.41(m,2h).
[0153]
化合物21i,1h nmr(600mhz,cdcl3)δ7.10(d,j=8.6hz,1h),6.92(d,j=8.2hz,2h),6.87(t,j=7.9hz,1h),6.83(d,j=8.4hz,1h),6.81(d,j=8.8hz,1h),6.76(dd,j=8.2,1.6hz,1h),6.61(s,1h),6.60(s,2h),6.44(dd,j=8.2,2.2hz,1h),6.23(d,j=7.5hz,1h),5.51(d,j=1.7hz,1h),4.14(t,j=5.9hz,2h),3.92(t,j=5.0hz,2h),3.89(s,3h),3.74
–
3.69(m,4h),3.53(s,4h),2.96(s,4h),2.81(t,j=5.8hz,4h),2.76
–
2.70(m,2h),2.57(s,4h),2.38(s,2h),2.24(s,4h).
[0154]
化合物21j和21k的制备:
[0155][0156]
参考化合物21f和21g的合成方法。以地钱素s和n
‑
(2
‑
氯乙基)吡咯烷盐酸盐为原料,得到化合物21j和21k两个白色固体。
[0157]
化合物21j,mp 75
‑
76℃。1h nmr(400mhz,cdcl3)δ7.07(d,j=8.5hz,1h),6.95(s,1h),6.93(s,1h),6.89(d,j=7.9hz,1h),6.85(s,1h),6.83(s,1h),6.76(dd,j=8.2,1.9hz,1h),6.63(d,j=2.3hz,2h),6.61(s,1h),6.49(dd,j=8.1,2.2hz,1h),6.24(d,j=7.5hz,1h),5.50(d,j=1.9hz,1h),3.90(s,3h),3.80
–
3.74(m,2h),2.99
–
2.94(m,2h),2.94
–
2.89(m,2h),2.82(dd,j=8.1,3.2hz,2h),2.78
–
2.72(m,2h),2.62(s,4h),2.60
–
2.54(m,2h),1.85(s,4h).
[0158]
化合物21k,mp 94
‑
95℃。1h nmr(600mhz,cd3od)δ7.31(d,j=8.8hz,1h),7.08(d,j
=8.8hz,1h),6.98(d,j=2.3hz,1h),6.97
–
6.95(m,2h),6.92(d,j=8.3hz,1h),6.81(dd,j=8.3,2.0hz,1h),6.64(s,1h),6.54(d,j=8.3hz,2h),6.52(dd,j=8.2,2.1hz,1h),6.33(d,j=7.5hz,1h),5.56(d,j=1.9hz,1h),4.41
–
4.37(m,2h),4.11
–
4.08(m,2h),3.84(s,3h),3.55(s,2h),3.34(d,j=15.7hz,4h),3.06
–
3.04(m,2h),3.00(d,j=9.2hz,4h),2.97(s,4h),2.84(d,j=5.7hz,2h),2.77(d,j=5.5hz,2h),2.07(t,j=6.6hz,4h),1.89
–
1.85(m,4h).
[0159]
实施例4化合物22a~c、27a~27g和27i~27k的合成
[0160]
化合物22a的制备:
[0161][0162]
将地钱素s(0.10mmol)溶解于1,4
‑
二氧六环(10ml)中,依次加入37%的甲醛(0.15mmol)、n
‑
苯基哌嗪(0.15mmol),回流搅拌12
‑
24h,真空浓缩溶液,通过硅胶柱层析纯化残余物,用meoh
‑
dcm溶液洗脱,得到棕色固体,mp 109
‑
110℃。1h nmr(400mhz,cdcl3)δ7.24(s,1h),7.22(d,j=4.8hz,1h),6.90(dd,j=9.7,4.3hz,5h),6.85(d,j=7.4hz,1h),6.80(d,j=8.1hz,1h),6.73(d,j=8.1hz,1h),6.64(s,1h),6.59(d,j=7.4hz,2h),6.56(s,1h),6.52(d,j=8.2hz,1h),6.24(d,j=7.3hz,1h),5.50(s,1h),3.86(s,3h),3.80(s,2h),3.23(s,4h),2.91(s,4h),2.87
–
2.65(m,8h).
[0163]
化合物22b的制备:
[0164]
参考化合物22a的合成方法,以地钱素s和1
‑
(4
‑
甲氧基)苯基哌嗪为原料,得到棕色固体,mp 119
‑
120℃。1h nmr(400mhz,cdcl3)δ6.96
–
6.93(m,2h),6.92(dd,j=5.7,3.2hz,3h),6.86(d,j=4.6hz,2h),6.84(d,j=3.6hz,1h),6.77(dd,j=8.2,1.9hz,1h),6.68(s,1h),6.65(s,1h),6.61(d,j=8.4hz,2h),6.55(dd,j=8.2,2.1hz,1h),6.28(d,j=7.5hz,1h),5.52(d,j=1.9hz,1h),3.90(s,3h),3.86(s,2h),3.78(s,3h),3.19(s,4h),2.95(s,4h),2.89
–
2.74(m,8h).
[0165][0166]
化合物22c的制备:
[0167][0168]
参考化合物22a的合成方法,以地钱素s和1
‑
(4
‑
溴)苯基哌嗪为原料,得到黄色固体,mp 219
‑
220℃。1h nmr(400mhz,cdcl3)δ7.36(d,j=8.8hz,2h),6.95(d,j=2.9hz,1h),6.93(t,j=3.7hz,2h),6.85(d,j=8.2hz,1h),6.81(s,1h),6.78(d,j=8.0hz,2h),6.68(s,1h),6.63(d,j=6.8hz,2h),6.60(s,1h),6.56(dd,j=8.3,2.4hz,1h),6.28(d,j=7.4hz,1h),5.54
–
5.51(m,1h),3.91(s,3h),3.83(s,2h),3.23(s,4h),2.95(s,4h),2.90
–
2.66(m,8h).
[0169]
化合物27a的制备:
[0170][0171]
参考化合物22a的合成方法,以地钱素s和二甲胺为原料,得到棕色固体,mp145
‑
146℃。1h nmr(400mhz,cdcl3)δ6.97
–
6.92(m,2h),6.91(s,1h),6.84(d,j=8.2hz,1h),6.77(dd,j=8.2,1.8hz,1h),6.67(s,1h),6.61(d,j=2.2hz,2h),6.59(s,1h),6.57
–
6.52(m,1h),6.28(d,j=7.4hz,1h),5.53(d,j=1.8hz,1h),3.90(s,3h),3.76(s,2h),2.94(s,4h),2.83(dd,j=8.3,3.3hz,2h),2.76(dd,j=8.2,3.0hz,2h),2.43(s,6h).
[0172]
化合物27b的制备:
[0173][0174]
参考化合物22a的合成方法,以地钱素s和二乙胺为原料,得到棕色固体,mp111
‑
112℃。1h nmr(400mhz,cdcl3)δ6.94(d,j=3.1hz,1h),6.91(d,j=3.8hz,1h),6.84(d,j=8.2hz,2h),6.79
–
6.74(m,1h),6.66(s,1h),6.61(d,j=9.0hz,2h),6.58(s,1h),6.57
–
6.53(m,1h),6.26(d,j=7.3hz,1h),5.52(d,j=1.7hz,1h),3.89(s,3h),3.88(s,2h),2.94(s,4h),2.82(d,j=7.4hz,2h),2.76(q,j=7.0hz,6h),1.19(t,j=7.1hz,6h).
[0175]
化合物27c的制备:
[0176][0177]
参考化合物22a的合成方法,以地钱素s和2
‑
氯
‑
n
‑
甲基乙胺盐酸盐为原料,得到白色固体,mp 64
‑
65℃。1h nmr(400mhz,cdcl3)δ6.97(d,j=6.6hz,2h),6.94(s,1h),6.86(d,j=8.2hz,1h),6.79(dd,j=8.2,1.7hz,1h),6.69(s,1h),6.64(d,j=3.6hz,2h),6.61(s,1h),6.59
–
6.55(m,1h),6.30(d,j=7.5hz,1h),5.56(d,j=1.8hz,1h),3.92(s,3h),3.85(s,2h),3.73(t,j=6.1hz,2h),2.97(s,4h),2.95(d,j=6.1hz,2h),2.86(dd,j=8.0,3.0hz,2h),2.79(dd,j=8.2,3.1hz,2h),2.42(s,3h).
[0178]
化合物27d的制备:
[0179][0180]
参考化合物22a的合成方法,以地钱素s和哌啶为原料,得到白色固体,mp184
‑
185℃。1h nmr(400mhz,cdcl3)δ6.94(d,j=3.7hz,1h),6.91(d,j=3.5hz,1h),6.84(d,j=8.2hz,2h),6.77(dd,j=8.2,1.9hz,1h),6.68(s,1h),6.60(d,j=8.4hz,2h),6.58(s,1h),
6.55(dd,j=8.2,2.2hz,1h),6.27(d,j=7.5hz,1h),5.53(d,j=1.9hz,1h),3.90(s,3h),3.75(s,2h),2.94(d,j=8.0hz,4h),2.89
–
2.40(m,8h),1.73
–
1.63(m,4h),1.63
–
1.44(m,2h).
[0181]
化合物27e的制备:
[0182][0183]
参考化合物22a的合成方法,以地钱素s和四氢吡咯为原料,得到黄色固体,mp208
‑
209℃。1h nmr(400mhz,cdcl3)δ6.91(d,j=8.1hz,2h),6.85
–
6.81(m,2h),6.76(dd,j=8.3,1.6hz,1h),6.64(s,1h),6.58(d,j=8.3hz,2h),6.52(d,j=7.9hz,1h),6.45(d,j=8.0hz,1h),6.30
–
6.23(m,1h),5.55
–
5.49(m,1h),3.89(s,3h),3.84(s,2h),2.95(d,j=18.5hz,4h),2.85
–
2.70(m,4h),2.01(d,j=23.8hz,4h),1.25(t,j=7.1hz,4h).
[0184]
化合物27f的制备:
[0185][0186]
参考化合物22a的合成方法,以地钱素s和n
‑
甲基哌嗪为原料,得到白色固体,mp110
‑
111℃。1h nmr(400mhz,cdcl3)δ6.94(d,j=6.8hz,2h),6.91(s,1h),6.84(d,j=8.2hz,1h),6.77(dd,j=8.2,1.8hz,1h),6.67(s,1h),6.63
–
6.60(m,2h),6.59(s,1h),6.54(dd,j=8.2,2.1hz,1h),6.28(d,j=7.5hz,1h),5.52(d,j=1.8hz,1h),3.90(s,3h),3.80(s,2h),3.00
–
2.60(m,16h),2.39(s,3h).
[0187]
化合物27g的制备:
[0188][0189]
参考化合物22a的合成方法,以地钱素s和吗啉为原料,得到白色固体,mp203
‑
204℃。1h nmr(400mhz,cdcl3)δ6.97
–
6.92(m,2h),6.92(s,1h),6.84(d,j=8.2hz,1h),6.77
(dd,j=8.2,1.7hz,1h),6.67(s,1h),6.61(s,2h),6.59(s,1h),6.54(dd,j=8.2,2.1hz,1h),6.28(d,j=7.5hz,1h),5.53(d,j=1.7hz,1h),3.90(s,3h),3.77(s,6h),2.94(s,4h),2.83(dd,j=8.4,3.0hz,2h),2.77(d,j=7.6hz,2h),2.64(s,4h).
[0190]
化合物27i的制备:
[0191][0192]
参考化合物22a的合成方法,以地钱素s和4
‑
羟基哌啶为原料,得到棕色固体,mp118
‑
119℃。1h nmr(400mhz,cdcl3)δ6.94(d,j=7.8hz,2h),6.91(s,1h),6.84(d,j=8.2hz,1h),6.77(dd,j=8.2,1.8hz,1h),6.67(s,1h),6.61(s,2h),6.59(s,1h),6.54(dd,j=8.3,2.1hz,1h),6.27(d,j=7.5hz,1h),5.52(d,j=1.8hz,1h),3.90(s,3h),3.81(s,2h),3.66
–
3.55(m,1h),2.94(s,6h),2.89
–
2.71(m,6h),2.06
–
1.98(m,2h),1.76
–
1.66(m,2h).
[0193]
化合物27j的制备:
[0194][0195]
参考化合物22a的合成方法,以地钱素s和n
‑
甲磺酰哌嗪为原料,得到白色固体,mp 228
‑
229℃。1h nmr(400mhz,cdcl3)δ6.94(d,j=8.6hz,2h),6.91(s,1h),6.84(d,j=8.2hz,1h),6.77(dd,j=8.2,1.7hz,1h),6.67(s,1h),6.63(s,1h),6.60(d,j=8.3hz,2h),6.53(dd,j=8.1,2.1hz,1h),6.29(d,j=7.5hz,1h),5.52(d,j=1.7hz,1h),3.90(s,3h),3.83(s,2h),3.34(s,4h),2.94(s,4h),2.86
–
2.74(m,11h).
[0196]
化合物27k的制备:
[0197]
[0198]
参考化合物22a的合成方法,以地钱素s和吗啉n
‑
甲磺酰哌嗪为原料,得到白色固体,mp 119
‑
120℃。1h nmr(600mhz,cdcl3)δ6.93(t,j=8.9hz,3h),6.84(d,j=8.2hz,1h),6.77(d,j=8.1hz,1h),6.68(s,1h),6.61(d,j=6.9hz,2h),6.59(s,1h),6.55
–
6.52(m,1h),6.27(d,j=7.4hz,1h),5.48(s,1h),3.90(s,3h),3.81(s,2h),3.76
–
3.72(m,4h),3.48
–
3.31(m,4h),3.28
–
3.22(m,4h),2.94(d,j=8.8hz,4h),2.85
–
2.80(m,2h),2.80
–
2.58(m,6h).
[0199]
实验例1化合物的抗肿瘤活性评价
[0200]
将地钱素s及其衍生物(按照实施例1
‑
4的方法制备)利用mtt法,测定对人肝癌细胞(hepg2)、人非小细胞肺癌细胞(a549)、人乳腺癌细胞(mcf
‑
7)、人前列腺癌细胞(pc3)和人支气管上皮样细胞(hbe)的抑制作用。
[0201]
方法:mtt法:取hepg2、a549、mcf
‑
7、pc3和hbe细胞株,2
×
104/孔细胞接种于96孔板中,分别加入0.4%dmso和不同浓度的化合物,培养48h后,每孔加入5mg/ml mtt20μl继续孵育4h,离心并小心吸取上清液,加入200μl dmso并轻微振荡,使生成的甲臜完全溶解显色后,用bio
‑
rad model 550microplate reader以570nm波长测od值。实验在不同培养时间条件下分别进行3次,算出平均值,具体结果见表1。
[0202]
表1地钱素s与衍生物的抗肿瘤活性
[0203][0204]
mtt筛选结果显示:在酚羟基修饰的衍生物中,化合物20对4种癌细胞均表现出显著的抑制作用,其半数抑制浓度ic
50
均小于12μm,对人正常细胞hbe细胞几乎无影响,化合物21a、21b、21d对hepg2、a549和mcf
‑
7这三种癌细胞表现出不错的抑制活性,对hbe细胞和pc3细胞的作用不明显,化合物21g、21j和21k对5种细胞均表现出非常显著的细胞毒性,尤其是
21g和21k作用非常显著,半数抑制浓度ic
50
均小于3μm。在胺甲基化修饰的衍生物中,化合物27i对hepg2和a549细胞表现出抑制活性,化合物27k对hepg2细胞表现出抑制活性。
[0205]
实验例2化合物的抗炎活性评价
[0206]
实验方法:将炎症细胞铺至6孔板内,加入10μm待测化合物溶液作用于细胞一定时间后,弃去培养基,以trizol法提取细胞的总rna,通过反转录试剂盒反转录成cdna后进行pcr扩增,利用软件计算相对转录水平(relative folds)。
[0207]
抗炎结果如表2中所示。
[0208]
表2地钱素s与衍生物的抗炎活性
[0209]
[0210][0211]
a
n/t means have not been tested.
[0212]
促炎因子白介素
‑
1β(il1β)转录水平的结果显示,衍生物中有8个化合物对il1β转录水平的影响表现出比地钱素s更好的抑制作用,其中化合物27c具有最显著的抑制效果,有9个化合物对il1β转录水平的影响与地钱素s相当。
[0213]
促炎因子白介素
‑
6(il6)转录水平的结果显示,衍生物中有11个化合物对il6转录水平的影响表现出比地钱素s更好的抑制作用,其中化合物20和27d具有最显著的抑制效果。
[0214]
综上,地钱素s及其衍生物能够显著抑制炎症因子il
‑
6和il
‑
1β的蛋白表达,并能够降低il
‑
6和il
‑
1β等炎症因子的基因表达水平,表现出良好的抗炎作用。此外,部分衍生物表现出良好的抗肿瘤活性。地钱素s及其衍生物均为具有良好开发前景的抗炎、抗风湿性关节炎和治疗由炎症引起肿瘤的新型药物。
[0215]
以上所述仅为本技术的优选实施例而已,并不用于限制本技术,尽管参照前述实施例对本技术进行了详细的说明,对于本领域的技术人员来说,其依然可以对前述各实施例所记载的技术方案进行修改,或者对其中部分技术特征进行等同替换。凡在本技术的精神和原则之内,所作的任何修改、等同替换、改进等,均应包含在本技术的保护范围之内。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1