一种吉美嘧啶中间体化合物的制作方法
1.本发明属于药物合成技术领域,具体涉及一种吉美嘧啶中间体化合物。
背景技术:
2.吉美嘧啶(gimeracil,cdhp),系5-氯-4-羟基-2(1h)-吡啶酮的通用名称,是日本taiho药品工业株式会社于1999年3月上市的口服抗肿瘤药cefecone(吉美嘧啶,替加氟,奥替拉西钾的组成摩尔比为1:0.4:1)的组分之一。替加氟是5-氟尿嘧啶的前药,口服吸收效果好,并在肝内逐渐转化为5-氟尿嘧啶,但5-氟尿嘧啶在体内极不稳定,易被肿瘤组织和正常器官产生的二氢嘧啶脱氢酶(dpd)快速降解失活。吉美嘧啶是一种dpd的可逆竞争性抑制剂,能高效抑制肿瘤组织中dpd,从而减慢5-氟尿嘧啶的分解速度,较长时间维持了5-氟尿嘧啶在血浆和肿瘤组织中的浓度,延长5-氟尿嘧啶的作用持续时间,从而增强了抗肿瘤活性,所以将5-氯-2,4-二羟基吡啶与替加氟联用能明显提高药物疗效。cefecone最初主要用来治疗肠癌和直肠癌,目前被认为是治疗晚期胃癌很有市场的抗肿瘤药之一。其中吉美嘧啶化学结构式为:
[0003][0004]
目前报道的关于吉美嘧啶的合成工艺主要有以下几种:
[0005]
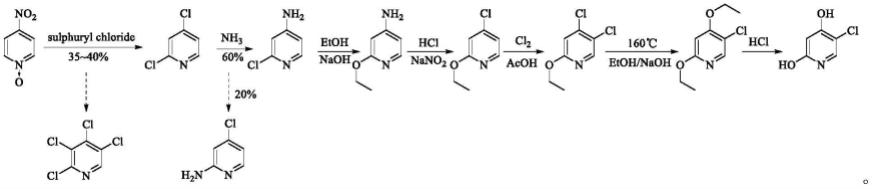
文献rec.tray.chim.,1953,72(2):285-295以4-硝基吡啶-n-氧化物为原料,经氯代、胺解、亲核取代、sandmeyer反应、氯代、亲核取代、酸解等7步反应而得,总收率12%。但该工艺操作繁琐,过程复杂,且前两步反应杂质较多,收率较低,且没有给出有效的精制方法,不适合工业化放大生产。具体合成路线如下:
[0006][0007]
文献rec.tray.chim.,1953,72(8):853-858通过得到3-氯-2,4-二羟基吡啶之后进行氯化水解,总收率26%。但该路线用到极为苛刻的封管反应条件,而且所用的氢溴酸是易腐蚀实验设备的试剂,使得该路线难以实现工业化生产。具体合成路线如下:
[0008][0009]
文献rec.tray.chim.,1953,73(4):704-708先得到2,4-二羟基吡啶之后再进行氯化水解,总收率25%,同样需要用到封管反应。具体合成路线如下:
[0010][0011]
日本专利特开平5-39241号公报由丙二酰氯为原料经3步反应得到吉莫斯特,该路线由于在中间体α,β-不饱和酮酸酯衍生物的合成原料的操作和反应条件上也或多或少地存在一些问题,使得该路线难以实现工业化生产。具体合成路线如下:
[0012][0013]
中国专利申请cn101107230a同族(wo2006080339、ep1842847)通过氯磺酸氯代后制得关键起始物料5-氯-1,3-二噁英-4-酮衍生物后与乙烯酮缩二乙醇衍生物反应得吡喃酮衍生物,最后用酸处理后再与氨反应进行环变换制得吉美嘧啶(以5-氯-2,2-二甲基-1,3-二噁英-4-酮计,总计收率为29.4%;以2,2-环己基-5-氯-1,3-二噁英-4-酮计,收率为53.3~72.4%)。但该路线起始物料需要自行制备,使得收率进一步降低,同时相关物料不易获得,生产成本较高,较难工业化放大生产。具体合成路线如下:
[0014][0015]
文献j.chem.res.,2018,vol.42,january,33-34以2,4-二甲氧基吡啶为起始物料,先经ncs氯代后,在水解,进而脱氯制得目标产品。具体合成路线如下:
[0016][0017]
而目前国内研究最多的是以文献archiv.der.pharmazie,1985,31:481-486为基础开发的以丙二腈、原乙酸三甲酯和n,n-二甲基甲酰胺二甲缩醛(dmf-dma)为起始原料先通过缩合反应制备1,1-二氰基-2-甲氧基-4-(n,n-二甲基氨基)-1,3-丁二烯,然后经体积分数(或质量分数)为80%的冰醋酸环合得3-氰基-4-甲氧基-2(1h)-吡啶酮,再经氯代形成5-氯-3-氰基-4-甲氧基-2(1h)-吡啶酮,最后经水解得吉美嘧啶。此外日本专利特开平5-78324、cn106316934a、cn112110854a及文献heterocycles.1993,36(1)145-148、吉莫斯特的合成.《沈阳药科大学学报》,2005,22(6),420-422、5-氯-2,4-二羟基吡啶的合成.《中国医药导报》,2007,4(20),14-15、二氢嘧啶脱氢酶抑制剂吉美拉西的合成.《华西药学杂志》,2008,23(3),252-254、5-氯-4-羟基-2-(1h)吡啶酮的合成.《化学试剂》,2008,30(12),939-940、吉美嘧啶及其关键中间体的制备.《齐鲁药事》,2012,31(3),132-133均采用该路线的
起始物料或关键中间体进行制备。相关反应路线如下:
[0018][0019]
由上可知,3-氰基-4-甲氧基-2(1h)-吡啶酮可用作制备吉美嘧啶的关键中间体,因此3-氰基4-甲氧基-2(1h)-吡啶酮直接影响该药品的生产、市场供应和质量问题。相关化合物结构式如下:
[0020][0021]
上述文献中,3-氰基4-甲氧基-2(1h)-吡啶酮的制备方法除文献二氢嘧啶脱氢酶抑制剂吉美拉西的合成.《华西药学杂志》,2008,23(3),252-254中将2-(1-甲氧基亚乙基)丙二腈单独分离制备外,其它文献均以丙二腈为起始物料,“一锅法”制得1,1-二氰基-2-甲氧基-4-(n,n-二甲基氨基)-1,3-丁二烯后再环化制备,但由于环化过程会生成胺臭味的沸点仅为9℃的二甲胺有毒气体,对环境危害较大;并且相关二甲胺气体进一步会与所用溶剂醋酸成盐析出,使得相关产品纯度较低,需要进一步水洗打浆除盐提纯,操作较为繁琐。
[0022]
鉴于目前制备3-氰基-4-甲氧基-2(1h)-吡啶酮时存在上述问题,研究寻找一条反应条件温和,操作过程简便,产品收率高、纯度高,生产成本低的适合工业化生产吉美嘧啶关键中间体3-氰基-4-甲氧基-2(1h)-吡啶酮的制备方法仍是目前需要解决的问题。
技术实现要素:
[0023]
为克服现有技术的缺陷,寻找一种更好的制备吉美嘧啶关键中间体3-氰基-4-甲氧基-2(1h)-吡啶酮的方法,本发提供了一种新的吉美嘧啶中间体化合物,及利用该新中间体制备3-氰基-4-甲氧基-2(1h)-吡啶酮的新方法,该方法所制得的目标产品具有较高的纯度以及收率,并且反应条件温和,操作过程简便,生产成本更低的特点。
[0024]
本发明的具体技术内容如下:
[0025]
本发明第一方面提供了一种新的吉美嘧啶中间体化合物,该化合物结构如式i-1所示:
[0026][0027]
本发明第二方面提供了该新吉美嘧啶中间体化合物的制备方法:一种吉美嘧啶中间体化合物i-1的制备方法包括以下步骤:
[0028]
室温,将化合物sm-1、化合物sm-2加入有机溶剂a中,控温至反应结束后,将反应液减压浓缩至干即为化合物i-1:
[0029][0030]
优选地,所述的化合物sm-1与化合物sm-2的投料摩尔比为1:1.0~2.0,优选1:1.2。
[0031]
优选地,所述的有机溶剂a选自1,4-二氧六环、四氢呋喃、乙腈、甲苯中的一种或其组合,其中特别优选1,4-二氧六环。
[0032]
优选地,所述的反应温度为60~100℃,优选80~85℃。
[0033]
本发明第三方面提供了该新中间体化合物i-1,用于制备吉美嘧啶关键中间体化合物3-氰基-4-甲氧基-2(1h)-吡啶酮的方法:
[0034][0035]
一种吉美嘧啶中间体化合物3-氰基-4-甲氧基-2(1h)-吡啶酮的制备方法,包括以下步骤:室温,将化合物i-1加入有机溶剂b中,控温回流至反应结束后,反应液降温析晶,将所得固体水洗,干燥后即为目标产品i。
[0036]
优选地,所述的有机溶剂b选自80%乙酸溶液、80%甲酸溶液中的一种或其组合,优选80%乙酸溶液。
[0037]
优选地,所述降温析晶温度为20~30℃。
[0038]
与现有技术相比,本发明取得的技术效果是:
[0039]
1.提供了一种新的吉美嘧啶中间体化合物,同时提供了利用该该新中间体简便高效的制备吉美嘧啶关键中间体3-氰基-4-甲氧基-2(1h)-吡啶酮的方法,整个合成方法操作简便,反应收率高;
[0040]
2.采用原甲酸三乙酯作为“一碳单位”供体,该试剂在环化过程中仅产生乙醇,可有效避免dmf-dma的使用,进而避免二甲胺气体或其醋酸盐的产生,使得反应操作更安全,更环保;
[0041]
3.通过本技术所得的3-氰基-4-甲氧基-2(1h)-吡啶酮具有较高的纯度与收率,适合工业化放大生产。
具体实施方式
[0042]
下面通过实施例来进一步说明本发明,应该正确理解的是:本发明的实施例仅仅是用于说明本发明,而不是对本发明的限制,所以,在本发明的方法前提下对本发明的简单改进均属于本发明要求保护的范围。
[0043]
对本发明得到的化合物i-1结构确证数据如下:
[0044][0045]
esi-hrms(m/z):179.0538[m+h]
+
;1h nmr(600mhz,dmso-d6)δ:6.85(d,j=11.4hz,1h),5.56(d,j=11.4hz,1h),4.46(q,j=7.8hz,2h),3.80(s,3h),1.45(t,j=7.8hz,3h);
13
c nmr(151mhz,dmso-d6)δ:180.63,158.46,112.46,98.76,65.47,63.49,57.55,13.87。
[0046]
对本发明得到的化合物i结构确证数据如下:
[0047][0048]
esi-hrms(m/z):191.1542[m+h]
+
;1h nmr(600mhz,dmso-d6)δ:12.13(s,1h),7.79(d,j=7.8hz,1h),6.35(d,j=7.8hz,1h),3.98(s,3h);
13
c nmr(151mhz,dmso-d6)δ:173.35,160.55,142.36,113.28,94.57,85.69,57.82。
[0049]
化合物i-1的合成
[0050]
实施例1
[0051]
室温,将2-(1-甲氧基亚乙基)丙二腈(sm-1,12.21g,0.10mol),原甲酸三乙酯(sm-2,17.78g,0.12mol)加入1,4-二氧六环(80ml)中,控温80~85℃反应,经检测反应完毕后,将反应液减压浓缩至干即为i-1,收率97.5%,hplc纯度99.92%。
[0052]
实施例2
[0053]
室温,将2-(1-甲氧基亚乙基)丙二腈(sm-1,12.21g,0.10mol),原甲酸三乙酯(sm-2,29.64g,0.20mol)加入甲苯(100ml)中,控温95~100℃反应,经检测反应完毕后,将反应液减压浓缩至干即为i-1,收率93.4%,hplc纯度99.64%。
[0054]
实施例3
[0055]
室温,将2-(1-甲氧基亚乙基)丙二腈(sm-1,12.21g,0.10mol),原甲酸三乙酯(sm-2,14.82g,0.10mol)加入四氢呋喃(60ml)中,控温60~65℃反应,经检测反应完毕后,将反应液减压浓缩至干即为i-1,收率92.5%,hplc纯度99.58%。
[0056]
实施例4
[0057]
室温,将2-(1-甲氧基亚乙基)丙二腈(sm-1,12.21g,0.10mol),原甲酸三乙酯(sm-2,32.61g,0.22mol)加入乙腈(60ml)中,控温55~60℃反应,经检测反应完毕后,将反应液减压浓缩至干即为i-1,收率88.5%,hplc纯度98.88%。
[0058]
化合物i的合成
[0059]
实施例5
[0060]
室温,将中间体i-1(8.91g,0.05mol)加入80%乙酸溶液(60ml)中,控温回流反应,经检测反应完毕后,反应液降至25℃析晶,有浅黄色针状结晶固体析出,过滤,将所得滤饼水洗,干燥后即为目标产品i,收率98.5%,hplc纯度99.95%。
[0061]
实施例6
[0062]
室温,将中间体i-1(8.91g,0.05mol)加入80%甲酸溶液(60ml)中,控温回流反应,经检测反应完毕后,反应液降至20℃析晶,有浅黄色针状结晶固体析出,过滤,将所得滤饼水洗,干燥后即为目标产品i,收率96.3%,hplc纯度99.68%。
[0063]
实施例7
[0064]
室温,将中间体i-1(8.91g,0.05mol)加入80%乙酸溶液(60ml)中,控温回流反应,经检测反应完毕后,反应液降至30℃析晶,有浅黄色针状结晶固体析出,过滤,将所得滤饼水洗,干燥后即为目标产品i,收率97.5%,hplc纯度99.72%。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1