一种苯并氮杂靛红化合物的合成的制作方法
1.本发明属于医药技术领域,具体的涉及一类结构新颖的苯并氮杂靛红中间体化合物及其制备方法。
背景技术:
2.靛红化合物在医药和有机光电材料领域受到广泛的关注。在医药研究领域,靛红及衍生物是一类重要的药物中间体和化学原料。有文献表明这类化合物的生物活性良好,在抗肿瘤、抗糖尿病、抗病毒、抗结核、抗菌、抗癫痫、杀虫等研究领域都有较好的应用。在有机光电材料领域,以靛红化合物为缺电子受体(a)及各种富电子给体(d)的d
‑
a型有机光电材料被广泛开发,并制备高性能的有机场效应晶体管和有机太阳能电池器件。
3.氮杂靛红(1h
‑
吡咯并[2,3
‑
c]吡啶
‑
2,3
‑
二酮)作为靛红的生物电子等排体,同样具有3位羰基活性位点,目前该系列的化合物合成、结构修饰及应用研究相对较少,而且这一结构中吡啶部分的修饰或替换研究仍未深入。因此,本文对氮杂靛红结构进行了修饰,设计合成了一苯并氮杂靛红药物中间体(1h
‑
吡咯并[2,3
‑
c]喹啉
‑
1,2(3h)
‑
二酮(化合物vi))。该结构具有较大的共轭体系,当用于合成有机光电材料时,有利于获得吸收光谱宽或电荷迁移率高等特点的有机共轭材料,当用于药物设计时,该方法改构有利于形成分子间相互作用,与生物体内的多种酶和受体相结合,提高生物活性,具有重大的潜在意义。
技术实现要素:
[0004]
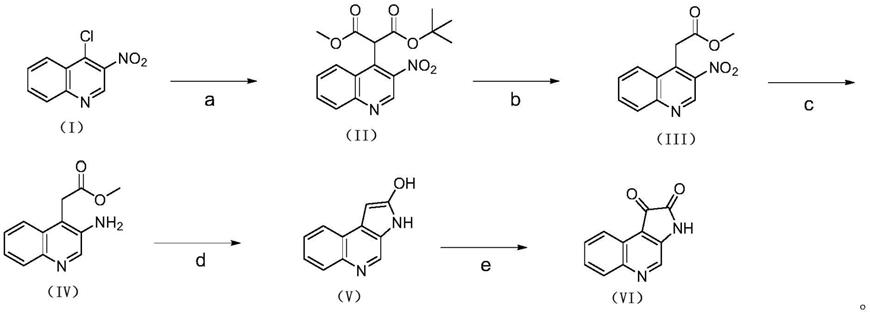
针对现有技术的不足,本发明目的在于提供一类结构新颖的苯并氮杂靛红中间体化合物,并且提供一种苯并氮杂靛红中间体的合成方法,该方法反应条件温和、步骤简单、经济适用,制备得到的苯并氮杂靛红中间体用途广泛,可以作为有机光电材料及医药领域药物中间体使用。
[0005]
具体来说,本发明提供一种式(vi)所示的化合物,或其互变异构体、立体异构体、溶剂化物、同位素衍生物或其药学上可接受的盐,其具有如下结构:
[0006][0007]
式(vi)所示的化合物的化学名为1h
‑
吡咯并[2,3
‑
c]喹啉
‑
1,2(3h)
‑
二酮,分子式为c
11
h6n2o2,分子量为199.13。
[0008]
本发明提供一种式(v)所示的化合物,或其互变异构体、立体异构体、溶剂化物、同位素衍生物或其药学上可接受的盐,其具有如下结构:
[0009][0010]
式(v)所示的化合物的化学名为3h
‑
吡咯并[2,3
‑
c]喹啉
‑2‑
醇,分子式为c
11
h8n2o,分子量为184.96。
[0011]
本发明提供一种式(iv)所示的化合物,或其互变异构体、立体异构体、溶剂化物、同位素衍生物或其药学上可接受的盐,其具有如下结构:
[0012][0013]
式(iv)所示的化合物的化学名为2
‑
(3
‑
氨基喹啉
‑4‑
基)乙酸甲酯,分子式为c
12
h
12
n2o2,分子量为216.94。
[0014]
本发明提供一种式(iii)所示的化合物,或其互变异构体、立体异构体、溶剂化物、同位素衍生物或其药学上可接受的盐,其具有如下结构:
[0015][0016]
式(iii)所示的化合物的化学名为2
‑
(3
‑
硝基喹啉
‑4‑
基)乙酸甲酯,分子式为c
12
h
10
n2o4,分子量为247.24。
[0017]
本发明提供一种式(ii)所示的化合物,或其互变异构体、立体异构体、溶剂化物、同位素衍生物或其药学上可接受的盐,其具有如下结构:
[0018][0019]
式(ii)所示的化合物的化学名为2
‑
(3
‑
硝基喹啉
‑4‑
基)丙二酸1
‑
(叔丁酯)3
‑
甲酯,分子式为c
17
h
18
n2o6,分子量为346.95。
[0020]
本发明进一步提供了上述式(vi)所示的化合物的合成方法。
[0021]
一种式(vi)所示的化合物的合成方法:包括如下步骤:
[0022][0023]
其中,所述式(vi)所示的化合物的合成方法具体包括以下步骤:
[0024]
步骤a:2
‑
(3
‑
硝基喹啉
‑4‑
基)丙二酸1
‑
(叔丁酯)3
‑
甲酯(ii)的制备
[0025]
将丙二酸叔丁酯甲酯溶于无水dmf中,冰浴条件下,加入nah,反应1h后,随后添加4
‑
氯
‑3‑
硝基喹啉,将所得混合物在室温条件搅拌4h,反应结束后,反应液倒入饱和氯化铵水溶液(30ml)淬灭,所得混合物用乙酸乙酯(3x 500ml)萃取,柱层析法纯化;
[0026]
步骤b:2
‑
(3
‑
硝基喹啉
‑4‑
基)乙酸甲酯(iii)的制备
[0027]
将上述2
‑
(3
‑
硝基喹啉
‑4‑
基)丙二酸1
‑
(叔丁酯)3
‑
甲酯(ii)溶于二氯甲烷中,加入三氟乙酸,室温反应3h,反应液减压浓缩,并将残余物用乙酸乙酯和水溶解,加入饱和碳酸氢钠溶液中和至中性,分离得到有机相,柱层析法纯化;
[0028]
步骤c:2
‑
(3
‑
氨基喹啉
‑4‑
基)乙酸甲酯(iv)的制备
[0029]
将上述2
‑
(3
‑
硝基喹啉
‑4‑
基)乙酸甲酯(iii)溶于乙醇中,加入催化剂,在氢气气氛下,升温搅拌,tlc监测原料完全反应后,反应溶液中加入二氯甲烷,过滤除去催化剂,减压蒸馏除去溶剂;
[0030]
步骤d:3h
‑
吡咯并[2,3
‑
c]喹啉
‑2‑
醇(v)的制备
[0031]
将上述2
‑
(3
‑
氨基喹啉
‑4‑
基)乙酸甲酯(iv)溶于thf,然后加浓盐酸,升温反应12h,tlc监测原料完全反应后,反应液减压除去溶剂,柱层析法纯化;
[0032]
步骤e:1h
‑
吡咯并[2,3
‑
c]喹啉
‑
1,2(3h)
‑
二酮(vi)的制备
[0033]
将上述3h
‑
吡咯并[2,3
‑
c]喹啉
‑2‑
醇(v)溶于dmso中,加入二氧化硒,升温反应1h,反应液加入饱和食盐水,用乙酸乙酯萃取,合并有机相,柱层析法纯化。
[0034]
优选地,本发明所述的1h
‑
吡咯并[2,3
‑
c]喹啉
‑
1,2(3h)
‑
二酮的合成方法,其中,步骤a中,丙二酸叔丁酯甲酯:nah:4
‑
氯
‑3‑
硝基喹啉的摩尔比为1:1.1:0.5。
[0035]
优选地,本发明所述的1h
‑
吡咯并[2,3
‑
c]喹啉
‑
1,2(3h)
‑
二酮的合成方法,其中,步骤b中,每1mmol的2
‑
(3
‑
硝基喹啉
‑4‑
基)丙二酸1
‑
(叔丁酯)3
‑
甲酯(ii)对应至少1.5ml的三氟乙酸。
[0036]
优选地,本发明所述的1h
‑
吡咯并[2,3
‑
c]喹啉
‑
1,2(3h)
‑
二酮的合成方法,其中,步骤c中,催化剂选自铁粉/氯化铵、氯化亚锡/浓盐酸、pd/c;进一步优选pd/c。
[0037]
优选地,本发明所述的1h
‑
吡咯并[2,3
‑
c]喹啉
‑
1,2(3h)
‑
二酮的合成方法,其中,步骤c中,每1mmol的2
‑
(3
‑
硝基喹啉
‑4‑
基)乙酸甲酯(iii)对应至少0.018g pd/c催化剂,反应温度控制在50~100℃;进一步优选反应温度控制在60~80℃;更进一步优选反应温度控制在60℃。
[0038]
优选地,本发明所述的1h
‑
吡咯并[2,3
‑
c]喹啉
‑
1,2(3h)
‑
二酮的合成方法,其中,步骤c中,在氢气气氛下,升温搅拌时间控制在2~6h;进一步优选升温搅拌时间控制在3~4h;更进一步优选升温搅拌时间控制在3h。
[0039]
优选地,本发明所述的1h
‑
吡咯并[2,3
‑
c]喹啉
‑
1,2(3h)
‑
二酮的合成方法,其中,步骤d中,2
‑
(3
‑
氨基喹啉
‑4‑
基)乙酸甲酯(iv)与浓盐酸的投料比至少1:10(n:n);进一步优选2
‑
(3
‑
氨基喹啉
‑4‑
基)乙酸甲酯(iv)与浓盐酸的投料比为1:10~1:15(n:n);更进一步优选2
‑
(3
‑
氨基喹啉
‑4‑
基)乙酸甲酯(iv)与浓盐酸的投料比为1:10(n:n)。
[0040]
优选地,本发明所述的1h
‑
吡咯并[2,3
‑
c]喹啉
‑
1,2(3h)
‑
二酮的合成方法,其中,步骤d中,反应温度控制在40~80℃;进一步优选反应温度为40~60℃;更进一步优选反应温度为40℃。
[0041]
优选地,本发明所述的1h
‑
吡咯并[2,3
‑
c]喹啉
‑
1,2(3h)
‑
二酮的合成方法,其中,步骤e中,3h
‑
吡咯并[2,3
‑
c]喹啉
‑2‑
醇(v)与二氧化硒的投料比为1:2~1:3(n:n);进一步优选3h
‑
吡咯并[2,3
‑
c]喹啉
‑2‑
醇(v)与二氧化硒的投料比为1:2(n:n)。
[0042]
优选地,本发明所述的1h
‑
吡咯并[2,3
‑
c]喹啉
‑
1,2(3h)
‑
二酮的合成方法,其中,步骤e中,反应温度控制在100~125℃;进一步优选反应温度为100℃。
[0043]
定义
[0044]
除另有规定外,n:n表示两个物料的摩尔比。
[0045]
除另有规定外,术语“药物上可接受的盐”是指在合理医学判断范围内适用于与哺乳动物特别是人的组织接触而无过度毒性、刺激、过敏反应等并与合理的效益/风险比相称的盐,比如胺、羧酸和其他类型化合物的医学上可接受的盐在所属领域中是被熟知的。可以在本发明化合物的最终分离和纯化期间原位制备所述盐,或单独通过将游离碱或游离酸与合适的试剂反应制备所述盐。
[0046]
除另有规定外,术语“同位素衍生物”是指本发明的化合物可以以同位素示踪的或富集形式存在,含有一个或多个原子,这些原子的原子量或质量数不同于自然界中发现的最大量的原子的原子量或质量数。同位素可以是放射性或非放射性的同位素。通常用作同位素标记的同位素是:氢同位素,2h和3h;碳同位素:
13
c和
14
c;氯同位素:
35
cl和
37
cl;氟同位素:
18
f;碘同位素:
123
i和
125
i;氮同位素:
13
n和
15
n;氧同位素:
15
o,
17
o和
18
o和硫同位素
35
s。这些同位素标记化合物可以用来研究药用分子在组织中的分布情况。尤其是2h和
13
c,由于它们容易标记且方便检测,运用更为广泛。某些重同位素,比如重氢(2h)的取代能增强代谢的稳定性,延长半衰期从而达到减少剂量的目的而提供疗效优势的。同位素标记的化合物一般从已被标记的起始物开始,用已知的合成技术象合成非同位素标记的化合物一样来完成其合成。
[0047]
除另有规定外,术语“溶剂化物”意指本发明化合物与一个或多个溶剂分子(无论有机的还是无机的)的物理缔合。该物理缔合包括氢键。在某些情形中,例如当一个或多个溶剂分子纳入结晶固体的晶格中时,溶剂化物将能够被分离。溶剂化物中的溶剂分子可按规则排列和/或无序排列存在。溶剂合物可包含化学计量或非化学计量的溶剂分子。“溶剂合物”涵盖溶液相和可分离的溶剂合物。示例性溶剂合物包括但不限于水合物、乙醇合物、甲醇合物和异丙醇合物。溶剂化方法是本领域公知的。
[0048]
除另有规定外,术语“立体异构体”是指具有相同化学构造,但原子或基团在空间
上排列方式不同的化合物。立体异构体包括对映异构体、非对映异构体、构象异构体(旋转异构体)、几何异构体(顺/反)异构体、阻转异构体等。所得的任何立体异构体的混合物可以依据组分物理化学性质上的差异被分离成纯的或基本纯的几何异构体,对映异构体,非对映异构体,例如,通过色谱法和/或分步结晶法。
[0049]
除另有规定外,术语“互变异构体”是指具有不同能量的可通过低能垒互相转化的结构异构体。若互变异构是可能的(如在溶液中),则可以达到互变异构体的化学平衡。例如,质子互变异构体(也称为质子转移互变异构体)包括通过质子迁移来进行的互相转化,如酮
‑
烯醇异构化和亚胺
‑
烯胺异构化。价键互变异构体包括通过一些成键电子的重组来进行的互相转化。
[0050]
除非其他方面表明,本发明所描述的结构式包括所有的同分异构形式(如对映异构,非对映异构,和几何异构(或构象异构)):例如含有不对称中心的r、s构型,双键的(z)、(e)异构体,和(z)、(e)的构象异构体。因此,本发明的化合物的单个立体化学异构体或其对映异构体,非对映异构体,或几何异构体(或构象异构体)的混合物都属于本发明的范围。
[0051]
制备实施例、实施例及本文其他地方使用的缩写词是:
[0052]
dmf
ꢀꢀꢀꢀꢀꢀꢀ
n,n
‑
二甲基甲酰胺
[0053]
thf
ꢀꢀꢀꢀꢀꢀꢀ
四氢呋喃
[0054]
dmso
ꢀꢀꢀ
二甲基亚砜
[0055]
本发明的有益效果为:
[0056]
1、本技术设计了一类结构新颖的苯并氮杂靛红中间体化合物,为制备靛红衍生物提供一个新的方法。
[0057]
2、本技术制备方法中步骤c的还原反应采用pd/c催化加氢还原硝基。发明人对比了铁粉/氯化铵的反应体系、氯化亚锡/浓盐酸反应体系、pd/c催化加氢反应体系,发现:铁粉反应过程中产生大量的氧化物,后处理较为麻烦;氯化亚锡/浓盐酸还原体系中浓盐酸的使用量通常比较大,且反应后处理需要消耗大量的碱,反应体系容易乳化,此外浓盐酸因其为易制毒类试剂,应用方面也有一定的限制;而pd/c虽价格昂贵,但pd/c作为一种高效、可回收、无污染的催化剂其优点也特别突出。为了实现高效硝基还原,采用控制变量法,以乙醇作为反应溶剂,发明人发现,步骤c反应过程的反应温度和反应时间控制对于产品收率和杂质水平控制有重要意义。发明人意外发现,步骤c控制反应温度在60~80℃,同时,反应时间控制在3~4h,对化合物(iv)收率及控制杂质生成有优良效果。
[0058]
3、本技术制备方法中对步骤d的关环反应条件进行优化,发明人发现,反应过程中化合物(iv)和浓盐酸的投料比以及反应温度对于产品收率和杂质水平控制有重要意义。发明人意外发现,化合物(iv)和浓盐酸的投料比在1:10~1:15(n:n),反应温度为40~60℃时,化合物(v)的收率最高,反应过程中产生的杂质最少。
[0059]
4、本技术制备方法中对步骤e的氧化反应条件进行优化,发明人意外发现,化合物(v)和二氧化硒的投料比在1:2~1:3(n:n),反应温度为100℃时,化合物(vi)的收率最高,反应过程中产生的杂质最少。
[0060]
5、本技术研究的合成方法反应条件温和,步骤简单,经济适用,利于规模化工业生产和应用。
具体实施方式
[0061]
以下结合具体实施例对本发明做进一步详细说明,但本发明并不限于以下实施例所提供的技术方案。应理解,这些实施例仅用于说明本发明而不用于限制本发明的范围。本领域技术人员可以借鉴本发明的内容,结合药物分析的相关原理,适当改进工艺参数来实现。特别需要指出的是,所有类似的替换和改动对本领域技术人员来说是显而易见的,它们都被视为包括在本发明范围内。
[0062]
为了更好地理解本发明而不是限制本发明的范围,在本技术中所用的表示用量、百分比的所有数字、以及其他数值,在所有情况下都应理解为以词语“大约”所修饰。各个数字参数至少应被看作是根据所报告的有效数字和通过常规的四舍五入方法而获得的。
[0063]
下述实施例中所使用的实验方法如无特殊说明,均为常规方法;下述实施例中所用的试剂、材料等,如无特殊说明,均可从商业途径得到。
[0064]
以下为本技术示例性化合物的制备实施例。
[0065]
实施例1
[0066][0067]
步骤a:2
‑
(3
‑
硝基喹啉
‑4‑
基)丙二酸1
‑
(叔丁酯)3
‑
甲酯(ii)的合成
[0068]
将丙二酸叔丁酯甲酯(3.48g,20mmol)溶于无水dmf(30ml)中,冰浴条件下,加入nah(0.88g,22mmol,60%w/w,分散于矿物油中),反应1h后,随后添加4
‑
氯
‑3‑
硝基喹啉(2.09g,10mmol)。将所得混合物在室温条件搅拌4h。反应结束后,反应液倒入饱和氯化铵水溶液(30ml)淬灭,所得混合物用乙酸乙酯(3x 500ml)萃取。将合并的有机相用盐水(3x 200ml)洗涤,经无水硫酸钠干燥并过滤,并在真空下浓缩,残余物柱层析纯化(v(正己烷):v(乙酸乙酯)=1:20~1:5)得到化合物(ii)(3.15g,收率91%)。
[0069]
esi
‑
ms(m/z):346.95[m+h]
+
;1h nmr(600mhz,dmso
‑
d6)δ:1.349(s,9h),3.680(s,3h),6.217(s,1h),7.867(t,j1=6.6hz,j2=9.0hz,1h),8.023(t,j1=6.6hz,j2=9.0hz,1h),8.220(d,j=8.4hz,1h),8.464(d,j=8.4hz,1h),9.435(s,1h)。
13
c nmr(150mhz,dmso
‑
d6)δ:166.63,164.38,149.27,142.72,136.10,133.16,130.41,129.55,126.86,126.15,83.39,53.30,51.39,27.78。
[0070]
步骤b:2
‑
(3
‑
硝基喹啉
‑4‑
基)乙酸甲酯(iii)的合成
[0071]
将上述2
‑
(3
‑
硝基喹啉
‑4‑
基)丙二酸1
‑
(叔丁酯)3
‑
甲酯(ii)(3.15g,9.1mmol)溶于二氯甲烷(20ml)中,加入三氟乙酸(15ml),室温反应3h,反应液减压浓缩,并将残余物用乙酸乙酯(300ml)和水(200ml)溶解,加入饱和碳酸氢钠溶液中和至中性。分离得到有机相,经无水硫酸钠干燥并过滤。将滤液减压浓缩并柱层析纯化(v(石油醚):v(乙酸乙酯)=1:20
~1:5)得到化合物(iii)(2.00g,收率89%)。
[0072]
esi
‑
ms(m/z):247.24[m+h]
+
;1h nmr(600mhz,dmso
‑
d6)δ:3.680(s,3h),4.587(s,2h),7.818(t,j1=7.8hz,j2=7.8hz,1h),7.983(t,j1=6.0hz,j2=7.8hz,1h),8.165(d,j=8.4hz,1h),8.452(d,j=8.4hz,1h),9.378(s,1h)。
13
c nmr(150mhz,dmso
‑
d6)δ:169.68,148.89,145.14,142.80,137.56,133.12,130.17,129.33,127.05,126.61,52.70,33.46。
[0073]
步骤c:2
‑
(3
‑
氨基喹啉
‑4‑
基)乙酸甲酯(iv)的合成
[0074]
将上述2
‑
(3
‑
硝基喹啉
‑4‑
基)乙酸甲酯(iii)(2.00g,8.1mmol)溶于乙醇(30ml)中,加入0.15g pd/c催化剂,在氢气气氛下,升温至60℃搅拌3h。tlc监测原料完全反应后,反应溶液中加入二氯甲烷,过滤除去催化剂,减压蒸馏除去溶剂,得到化合物(iv)(1.67g,收率96%)。
[0075]
esi
‑
ms(m/z):216.94[m+h]
+
;1h nmr(600mhz,dmso
‑
d6)δ:3.614(s,3h),4.017(s,2h),5.676(s,2h),7.361(t,j1=7.2hz,j2=7.8hz,1h),7.441(t,j1=7.2hz,j2=7.8hz,1h),7.691(d,j=7.2hz,1h),7.816(d,j=7.8hz,1h),8.512(s,1h)。
13
c nmr(150mhz,dmso
‑
d6)δ:171.35,144.35,141.75,141.01,129.72,129.02,127.18,124.16,122.27,114.58,52.26,31.18。
[0076]
步骤d:3h
‑
吡咯并[2,3
‑
c]喹啉
‑2‑
醇(v)的合成
[0077]
将上述2
‑
(3
‑
氨基喹啉
‑4‑
基)乙酸甲酯(iv)(1.18g,5.7mmol)溶于thf(30ml),然后加浓盐酸(4.8ml,57mmol),升温至40℃反应12h,tlc监测原料完全反应后,反应液减压除去溶剂,残渣通过柱层析纯化(v(乙酸乙酯):v(正己烷)=1:1~4:1)得到化合物(v)(0.93g,收率93%)。
[0078]
esi
‑
ms(m/z):184.96[m+h]
+
;1h nmr(600mhz,dmso
‑
d6)δ:5.861(s,1h),7.299(t,j1=7.2hz,j2=7.2hz,1h),7.499(t,j1=7.2hz,j2=7.2hz,1h),7.544(s,1h),7.650(d,j=7.2hz,1h),8.003(d,j=7.8hz,1h),10.330(s,1h),12.200(s,1h)。
13
c nmr(150mhz,dmso
‑
d6)δ:170.95,138.14,134.46,128.81,125.66,123.16,123.05,118.76,116.51,112.99,91.79。
[0079]
步骤e:1h
‑
吡咯并[2,3
‑
c]喹啉
‑
1,2(3h)
‑
二酮(vi)的合成
[0080]
将上述3h
‑
吡咯并[2,3
‑
c]喹啉
‑2‑
醇(v)(0.57g,3.1mmol)溶于dmso(10ml)中,加入二氧化硒(0.70g,6.2mmol),升温至100℃反应1h,反应液加入饱和食盐水(20ml),用乙酸乙酯(3x 500ml)萃取,合并有机相,并柱层析纯化(v(二氯甲烷):v(甲醇)=50:1~10:1)得到化合物(vi)(0.61g,收率75%)。
[0081]
esi
‑
ms(m/z):199.13[m+h]
+
;1h nmr(600mhz,dmso
‑
d6)δ:7.662(t,j1=7.8hz,j2=7.2hz,1h),7.738(t,j1=7.8hz,j2=7.2hz,1h),8.023(d,j=8.4hz,1h),8.314(d,j=8.4hz,1h),8.822(s,1h),11.255(s,1h)。
13
c nmr(150mhz,dmso
‑
d6)δ:185.05,159.58,146.42,145.63,139.57,131.18,130.32,128.15,122.67,121.46,114.04。
[0082]
以下是本发明的对比例。
[0083]
对比例1:
[0084]
本对比例步骤c中以不同反应温度替代实施例1步骤c中的60℃,其他反应条件与实施例1相同。结果见表1。
[0085]
表1反应温度对化合物(iv)收率的影响
[0086]
反应温度/℃2540506080100110收率/%12507296949190
[0087]
对比例2:
[0088]
本对比例步骤c中以不同反应时间替代实施例1步骤c中的3h,其他反应条件与实施例1相同。结果见表2。
[0089]
表2反应时间对化合物(iv)收率的影响
[0090]
反应时间/h0.5123456收率/%25608196929088
[0091]
对比例3:
[0092]
本对比例步骤d中以不同的化合物(iv)和浓盐酸的投料比替代实施例1步骤d中的1:10(n:n),其他反应条件与实施例1相同。结果见表3。
[0093]
表3投料比对化合物(v)收率的影响
[0094]
投料比/n
(化合物iv)
∶n
(浓盐酸)
1:11:51:101:121:15收率/%3064939293
[0095]
对比例4:
[0096]
本对比例步骤d中以不同的反应温度替代实施例1步骤d中的40℃,其他反应条件与实施例1相同。结果见表4。
[0097]
表4反应温度对化合物(v)收率的影响
[0098]
反应温度/℃25406080收率/%50938782
[0099]
对比例5:
[0100]
本对比例步骤e中以不同的化合物(v)和二氧化硒的投料比替代实施例1步骤e中的1:2(n:n),其他反应条件与实施例1相同。结果见表5。
[0101]
表5投料比对化合物(vi)收率的影响
[0102]
投料比/n
(化合物v)
∶n
(二氧化硒)
1:11:21:3收率/%417573
[0103]
对比例6:
[0104]
本对比例步骤e中以不同的反应温度替代实施例1步骤e中的100℃,其他反应条件与实施例1相同。结果见表6。
[0105]
表6反应温度对化合物(vi)收率的影响
[0106][0107][0108]
以上对比数据表明,以4
‑
氯
‑3‑
硝基喹啉为原料,经取代、脱boc、还原、关环和氧化,合成了中间体1h
‑
吡咯并[2,3
‑
c]喹啉
‑
1,2(3h)
‑
二酮(vi)。发明人对合成路线中主要的关键步骤进行了优化,研究了不同反应温度、反应时间和投料比等影响因素,得到了如下意外发现:步骤c中反应温度60~80℃,反应时间为3~4h,此时产物(iv)收率为96%;步骤d中
n(化合物(iv)):n(浓盐酸)=1:10~1:15,反应温度为40~60℃,此时产物(v)收率为93%;步骤e中n(化合物(v)):n(二氧化硒)=1:2~1:3,反应温度为100℃,此时产物(vi)收率为75%。经五步反应得到化合物(vi)的总收率为54.23%。综上,上述优化条件对各产物收率及控制杂质生成具有优良效果。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1