一种铱催化的N-苯基-7氮杂吲哚衍生物及其制备方法
一种铱催化的n
‑
苯基
‑
7氮杂吲哚衍生物及其制备方法
技术领域
1.本发明属于精细有机合成技术领域,具体为一种铱催化的n
‑
苯基
‑
7氮杂吲哚衍生物及其制备方法。
背景技术:
2.吲哚骨架是一类相当重要的碳环结构,并且由于在许多活性生物大分子中广泛存在,因此能在许多药剂中发现引哚骨架。7
‑
氮杂吲哚是由一个氮原子取代7位吲哚的碳原子所形成的杂环,与吲哚互为等电子体,7
‑
氮杂吲哚作为氮杂吲哚家族中的关键一员,其骨架广泛存在于活性天然产物及药物分子的核心结构单元中,研究表明该类化合物具有抗癌、抗菌、抗糖尿病等重要的生物活性。因此药物化学家常以含7
‑
氮杂吲哚骨架的化合物为先导化合物对其进行结构改造与修饰,通过分析与靶点的构效关系来寻找疗效更加理想的药物。
3.目前,有关7
‑
氮杂吲哚类化合物的应用属于热点,但有关以7
‑
氮杂吲哚为导向基团,实现n
‑
芳基上的c
‑
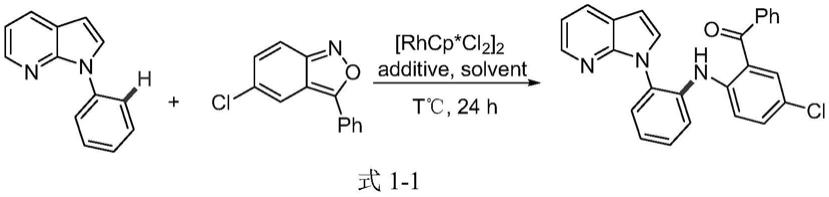
h胺基化反应却仍然很少,主要有两个反应,一个是2017年,kim课题组报道了如式1
‑
1所示的铑催化的7
‑
氮杂吲哚的胺基化反应,在该反应中苯并异噻唑作为胺源,实现了c
‑
n键的偶联(见文献“mijin jeon,jihye,et al.adv.synth.catal.2017,359,3471
‑
3478”)。另一个是2018年,董课题组报道了如式1
‑
2所示的以芳基叠氮为胺源,开发了铱催化的n
‑
芳基
‑7‑
氮杂吲哚的c
‑
n胺基化反应(见文献“w.
‑
h.li,l.dong,et.al.,adv.synth.catal,2018,360,1104.”)。
[0004][0005]
[0006]
但是,目前以7
‑
氮杂吲哚为导向基团的n
‑
芳基
‑7‑
氮杂吲哚c
‑
h胺基化反应,来制备n
‑
芳基
‑
7氮杂吲哚衍生物时,反应过程时间较长,转化率低。
技术实现要素:
[0007]
为了克服上述现有技术的不足,本发明提供一种铱催化的n
‑
苯基
‑
7氮杂吲哚衍生物及其制备方法,得到了n
‑
苯基
‑
7氮杂吲哚衍生物,廉价,高效,操作简单,转化率高,选择性好。
[0008]
本发明是通过如下技术方案来实现:
[0009]
一种铱催化的n
‑
苯基
‑
7氮杂吲哚衍生物,该n
‑
苯基
‑
7氮杂吲哚衍生物的结构式如下所示:
[0010][0011]
其中,r1=h、br、cl或ome,r2=h、me、f或co2me。
[0012]
优选的,包括如下步骤:
[0013]
将n
‑
苯基
‑
7氮杂吲哚和酰氧基胺基甲酸酯类化合物在二氯(五甲基环戊二烯基)合铱(iii)二聚体、五甲基环戊二烯醋酸铱、二氯双(五甲基环戊二烯)二氯化铱或(乙腈基)环戊二烯六氟磷酸铱的催化下进行7
‑
氮杂吲哚n
‑
苯基上的c
‑
h胺基化反应,得到反应物,之后分离产物得到n
‑
苯基
‑
7氮杂吲哚衍生物。
[0014]
进一步,具体包括如下步骤:
[0015]
将n
‑
苯基
‑
7氮杂吲哚、酰氧基胺基甲酸酯类化合物、离子液体和催化剂混合均匀,催化剂为二氯(五甲基环戊二烯基)合铱(iii)二聚体、五甲基环戊二烯醋酸铱、二氯双(五甲基环戊二烯)二氯化铱或(乙腈基)环戊二烯六氟磷酸铱,n
‑
苯基
‑
7氮杂吲哚和酰氧基胺基甲酸酯类化合物的摩尔比为1:1.5,得到混合体系;
[0016]
将混合体系在25~30℃下进行反应,得到反应液;
[0017]
将反应液中的产物依次进行分离、提纯,得到n
‑
苯基
‑
7氮杂吲哚衍生物。
[0018]
优选的,将n
‑
苯基
‑
7氮杂吲哚、酰氧基胺基甲酸酯类化合物、离子液体、添加剂和催化剂混合均匀,添加剂为六氟锑酸银、三氟甲磺酸银或三氟甲烷磺酸铵银,得到混合体系。
[0019]
进一步,所述催化剂的摩尔数占n
‑
苯基
‑
7氮杂吲哚、酰氧基胺基甲酸酯类化合物、添加剂和催化剂总摩尔数的5%,催化剂和添加剂的摩尔比为1:4。
[0020]
优选的,所述的离子液体为1
‑
丁基
‑3‑
甲基咪唑四氟硼酸盐、1
‑
丁基
‑3‑
甲基咪唑二(三氟甲基磺酰)酰亚胺或1
‑
丁基
‑3‑
甲基咪唑六氟磷酸盐。
[0021]
优选的,所述的n
‑
苯基
‑
7氮杂吲哚为n
‑
(3
‑
甲基苯)
‑7‑
氮杂吲哚、n
‑
(4
‑
氟化苯)
‑
7
‑
氮杂吲哚、n
‑
(4
‑
甲氧羰基苯)
‑7‑
氮杂吲哚、n
‑
苯基
‑4‑
氯
‑7‑
氮杂吲哚或n
‑
苯基
‑5‑
溴
‑7‑
氮杂吲哚。
[0022]
优选的,所述的混合体系在25~30℃下反应12~36h,得到反应液。
[0023]
优选的,将反应液先用环己烷进行萃取,得到萃取液,之后将萃取液中的环己烷通过旋转蒸发除去,所得的残留物用柱层析色谱进行提纯,得到n
‑
苯基
‑
7氮杂吲哚衍生物。
[0024]
进一步,将残留物用柱层析色谱进行提纯时,依次以石油醚:乙酸乙酯=30:1、石油醚:乙酸乙酯=20:1、石油醚:乙酸乙酯=10:1、石油醚:乙酸乙酯=5:1和石油醚:乙酸乙酯=3::1的混合液作为洗脱剂进行洗脱,得到n
‑
苯基
‑
7氮杂吲哚衍生物。
[0025]
与现有技术相比,本发明具有以下有益的技术效果:
[0026]
本发明一种铱催化的n
‑
苯基
‑
7氮杂吲哚的制备方法,以n
‑
苯基
‑
7氮杂吲哚作为主反应物,利用主反应物中7
‑
氮杂吲哚的导向定位作用,再以酰氧基胺基甲酸酯类化合物为胺源,通过催化剂二氯(五甲基环戊二烯基)合铱(iii)二聚体、五甲基环戊二烯醋酸铱、二氯双(五甲基环戊二烯)二氯化铱或(乙腈基)环戊二烯六氟磷酸铱,可以在一定程度上降低c
‑
h键的活化能垒,从而提高其反应活性,选择性好,转化率高,制备了7
‑
氮杂吲哚n
‑
苯基上的c
‑
h胺基化反应产物,得到了一种全新的n
‑
苯基
‑
7氮杂吲哚衍生物。本发明实现了高效催化下的7
‑
氮杂吲哚n
‑
芳基上的c
‑
h胺基化反应,为药物化学家寻找有良好价值的7
‑
氮杂吲哚的衍生物提供一种新途径,具有广阔的应用前景。
[0027]
进一步的,添加剂为银盐,可提高催化性能,并且在胺化反应中,离子液体具有反应速率比普通溶剂中快几倍的优势且离子液体在反应过程中起到了溶剂和催化剂的双重作用,这样通过催化剂、离子液体以及添加剂高效的催化活性可以进一步降低c
‑
h键的活化能垒,从而提高反应活性。
具体实施方式
[0028]
为了加深对本发明的理解,下面将结合实施例对本发明做进一步的说明,该实施例仅用于解释本发明,并不构成对本发明的保护范围的限定。
[0029]
本发明实施例涉及到的材料、试剂和实验设备,如无特别说明,均为符合有机化合物合成领域的市售产品。
[0030]
本发明一种铱催化的n
‑
苯基
‑
7氮杂吲哚衍生物的制备方法,包括如下步骤:
[0031]
在15ml耐压管里加入1当量(0.05mmol)n
‑
苯基
‑
7氮杂吲哚,1.5当量的胺基化试剂(酰氧基胺基甲酸酯类化合物),5mol%催化剂(含铱的配合物)、20mol%银盐和1ml溶剂(离子液体),在25~30℃和常压(0.1mpa)下搅拌反应12~36h,再通过薄层色谱分离提纯,即可得到目标产物。
[0032]
反应式如下所示:
[0033][0034]
其中,r1=h、br、cl或ome,r2=h、me、f或co2me,反应主要发生在苯基的邻位,所以选择性好。
[0035]
n
‑
苯基7
‑
氮杂吲哚为n
‑
(3
‑
甲基苯)
‑7‑
氮杂吲哚、n
‑
(4
‑
氟化苯)
‑7‑
氮杂吲哚、n
‑
(4
‑
甲氧羰基苯)
‑7‑
氮杂吲哚、n
‑
苯基
‑4‑
氯
‑7‑
氮杂吲哚或n
‑
苯基
‑5‑
溴
‑7‑
氮杂吲哚。
[0036]
溶剂为bmimbf4、bmimntf2或bmimpf6。银盐作为添加剂可提高其催化性能,具体为六氟锑酸银(agsbf6)、三氟甲磺酸银(agotf)、三氟甲烷磺酸铵银(agntf2)中的一种。
[0037]
实施例1:
[0038][0039]
在室温条件下,依次准确称量n
‑
(3
‑
甲基苯)
‑7‑
氮杂吲哚0.0104g(0.05mmol),((3
‑
甲基苯甲酰基)氧基)氨基甲酸叔丁酯(氨基化试剂)0.0188g(0.075mmol),二氯(五甲基环戊二烯基)合铱(iii)二聚体([ircp*cl2]2)(5mol%),agntf2(20mol%),1
‑
丁基
‑3‑
甲基咪唑四氟硼酸盐(bmimbf4,1ml);
[0040]
将测量好的上述物质依次加入装有磁子的25ml耐压管中,封闭耐压管,室温条件下在磁力搅拌器上搅拌,然后缓慢加热至30℃。在0.1mpa下,12h后停止反应,冷却至室温,将混合物用10ml的环己烷萃取3次,合并有机相,利用旋转蒸发仪将多余的环己烷除去,残留物用柱层析色谱分离,以300~400目的硅胶作为固定相,依次以石油醚:乙酸乙酯=30:1;石油醚:乙酸乙酯=20:1;石油醚:乙酸乙酯=10:1;石油醚:乙酸乙酯=5:1;石油醚:乙酸乙酯=3:1的乙酸乙酯和石油醚的混合溶剂作为洗脱剂对产品进行纯化分离,产率为80%。
[0041]
离子液体具有不挥发、可循环使用、化学可修饰性强等优点,在分离过程、均相催化、生物化工等领域有广泛的应用。
[0042]
所得(4
‑
甲基
‑2‑
(1h
‑
吡咯并[2,3
‑
b]吡啶
‑1‑
基)苯基)氨基甲酸叔丁酯的表征数据如下:
[0043]1h nmr(400mhz,chloroform
‑
d)δ8.40(d,j=4.7hz,1h),8.04(s,1h),7.94(d,j=8.2hz,1h),7.48(s,1h),7.39(d,j=3.6hz,1h),7.27(d,j=8.4hz,1h),7.20(dd,j=7.8,4.8hz,1h),7.11(s,1h),6.72(d,j=3.6hz,1h),2.40(s,3h)ppm.
[0044]
13
c nmr(101mhz,chloroform
‑
d)δ153.46,148.06,143.67,134.29,131.41,129.72,129.53,129.16,127.96,121.32,116.67,102.34,80.22,28.26,20.73ppm.
[0045]
核磁数据表明所得产物与预期一致。
[0046]
实施例2:
[0047][0048]
在室温条件下,依次准确称量n
‑
(4
‑
氟化苯)
‑7‑
氮杂吲哚0.0106g(0.05mmol),氨基化试剂0.0188g(0.075mmol),五甲基环戊二烯醋酸铱(ircp*(oac)2)(5mol%),agsbf6(20mol%),1
‑
丁基
‑3‑
甲基咪唑二(三氟甲基磺酰)酰亚胺(bmimntf2,1ml);
[0049]
将测量好的上述物质依次加入装有磁子的25ml耐压管中,封闭耐压管,室温条件下在磁力搅拌器上搅拌,然后缓慢加热至30℃。在0.1mpa下,12h后停止反应,冷却至室温,纯化同(实施例1),产率为92%。
[0050]
所得(5
‑
氟
‑2‑
(1h
‑
吡咯并[2,3
‑
b]吡啶
‑1‑
基)苯基)氨基甲酸叔丁酯表征数据如下:
[0051]1h nmr(400mhz,chloroform
‑
d)δ21.50(s,1h),8.39(dd,j=4.7,1.6hz,1h),8.07(dd,j=7.8,1.6hz,1h),8.00(dd,j=11.0,2.9hz,1h),7.44(s,1h),7.33(d,j=3.6hz,1h),7.23(ddd,j=8.1,5.2,3.5hz,2h),6.90(ddd,j=8.8,7.5,2.9hz,1h),6.75(d,j=3.6hz,1h),1.31(s,9h)ppm.
[0052]
13
c nmr(101mhz,chloroform
‑
d)δ152.64,148.18,143.83,136.20,129.80,129.56,128.96,128.86,124.36,121.22,116.90,110.56,110.33,109.62,109.34,102.72,81.00,29.68,28.17ppm.
[0053]
核磁数据表明所得产物与预期一致。
[0054]
实施例3
[0055][0056]
在室温条件下,依次准确称量n
‑
(4
‑
甲氧羰基苯)
‑7‑
氮杂吲哚0.0126g(0.05mmol),氨基化试剂0.0188g(0.075mmol),(乙腈基)环戊二烯六氟磷酸铱([cp*ir(ch3cn)3](sbf6)2)(5mol%),agotf(20mol%),1
‑
丁基
‑3‑
甲基咪唑四氟硼酸盐(bmimbf4,1ml);
[0057]
将测量好的上述物质依次加入装有磁子的25ml耐压管中,封闭耐压管,室温条件下在磁力搅拌器上搅拌,然后缓慢加热至30℃。在0.1mpa下,12h后停止反应,冷却至室温,纯化同(实施例1),产率为60%。
[0058]
所得3
‑
((叔丁氧基羰基)氨基)
‑4‑
(1h
‑
吡咯并[2,3
‑
b]吡啶
‑1‑
基)苯甲酸酯的表征数据如下:
[0059]1h nmr(400mhz,chloroform
‑
d)δ8.75(d,j=2.1hz,1h),8.42(dd,j=4.7,1.7hz,1h),8.09(dd,j=7.8,1.6hz,1h),8.01(s,1h),7.93(dd,j=8.3,2.0hz,1h),7.45
–
7.35(m,2h),7.25(dd,j=7.8,4.8hz,1h),6.79(d,j=3.6hz,1h),4.01(s,2h),1.49(s,9h)ppm.
[0060]
13
c nmr(101mhz,chloroform
‑
d)δ166.38,153.12,148.03,143.71,133.88,133.58,130.05,129.94,129.36,127.33,125.48,125.39,121.61,117.08,103.45,80.73,52.34,28.21ppm.
[0061]
核磁数据表明所得产物与预期一致。
[0062]
实施例4
[0063][0064]
在室温条件下,依次准确称量n
‑
苯基
‑4‑
氯
‑7‑
氮杂吲哚0.0114g(0.05mmol),氨基化试剂0.0188g(0.075mmol),二氯双(五甲基环戊二烯)二氯化铱([ircp*cl2]2)(5mol%),agsbf6(20mol%),1
‑
丁基
‑3‑
甲基咪唑六氟磷酸盐(bmimpf6,1ml);
[0065]
将测量好的上述物质依次加入装有磁子的25ml耐压管中,封闭耐压管,室温条件下在磁力搅拌器上搅拌,然后缓慢加热至30℃。在0.1mpa下,12h后停止反应,冷却至室温,纯化同(实施例1),产率为91%。
[0066]
所得(2
‑
(4
‑
氯
‑
1h
‑
吡咯并[2,3
‑
b]吡啶
‑1‑
基)苯基)氨基甲酸叔丁酯的表征数据如下:
[0067]1h nmr(400mhz,chloroform
‑
d)δ8.28(d,j=5.2hz,1h),8.09(d,j=8.3hz,1h),7.52
–
7.36(m,4h),7.33
–
7.23(m,2h),7.24(dd,j=5.4,1.6hz,2h),6.85(d,j=3.6hz,1h),1.31(s,9h),ppm.
[0068]
13
c nmr(101mhz,chloroform
‑
d)δ153.15,148.52,144.04,136.92,134.11,130.18,129.01,128.88,127.60,124.33,123.79,120.61,116.98,100.98,80.59,29.70,28.23ppm.
[0069]
核磁数据表明所得产物与预期一致。
[0070]
实施例5
[0071][0072]
在室温条件下,依次准确称量n
‑
苯基
‑5‑
溴
‑7‑
氮杂吲哚0.0136g(0.05mmol),氨基化试剂0.0188g(0.075mmol),五甲基环戊二烯醋酸铱(ircp*(oac)2)(5mol%),agotf(20mol%),1
‑
丁基
‑3‑
甲基咪唑二(三氟甲基磺酰)酰亚胺(bmimntf2,1ml);
[0073]
将测量好的上述物质依次加入装有磁子的25ml耐压管中,封闭耐压管,室温条件下在磁力搅拌器上搅拌,然后缓慢加热至30℃。在0.1mpa下,12h后停止反应,冷却至室温,
纯化同(实施例1),产率为85%。
[0074]
所得(2
‑
(4
‑
氯
‑
1h
‑
吡咯并[2,3
‑
b]吡啶
‑1‑
基)苯基)氨基甲酸叔丁酯的表征数据如下:
[0075]1h nmr(400mhz,chloroform
‑
d)δ8.39(d,j=2.2hz,3h),8.17(d,j=2.1hz,3h),8.08(d,j=8.3hz,3h),7.47(ddd,j=8.6,6.9,2.0hz,3h),7.39(d,j=3.6hz,3h),7.33
–
7.23(m,5h),7.27
–
7.19(m,5h),6.69(d,j=3.6hz,3h),2.65(s,0h),1.32(s,2h),1.30(s,9h)ppm.
[0076]
13
c nmr(101mhz,chloroform
‑
d)δ153.12,146.45,144.22,134.16,131.73,131.13,128.88,128.81,127.62,124.30,123.66,122.82,112.71,101.98,80.66,29.69,28.23ppm.
[0077]
核磁数据表明所得产物与预期一致。
[0078]
以上所述,仅为本发明的优选实施例,应当指出,对于本技术领域的普通技术人员来讲,在不脱离本发明的核心技术的前提下,还可以做出改进和润饰,这些改进何润饰也应当属于本发明的专利保护范围。与本发明的权利要求书相当的含义和范围内的任何改变,都应认为是包括在权利要求书范围之内的。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1