1500nm以上成像的有机荧光小分子化合物及其制备方法和应用
文档序号:32785370发布日期:2023-01-03 18:24阅读:74来源:国知局
导航: X技术> 最新专利>有机化合物处理,合成应用技术
1.本发明专利涉及生物医学荧光成像应用技术领域,尤其是指1500nm以上成像的有机荧光小分子化合物及其制备方法和应用。
背景技术:
2.组织缺血性疾病需要及时和早期诊断缺血部位的外科机械血栓切除术或药物溶栓。以缺血性中风或下肢动静脉血栓形成为代表的血管疾病,如果不及时治疗,会导致患者残疾和行为语言障碍,甚至危及生命。因此,准确、动态地显示缺血部位有助于及时抑制疾病的进一步恶化。然而,传统的超声成像受信号衰减、不可避免的组织信号干扰和电离辐射等因素的限制,难以实现深部组织的血管造影,严重依赖于声音与体内组织的物理相互作用。尽管计算机断层扫描(ct)、磁共振成像(mri)和正电子发射断层扫描(pet)的良好穿透深度已被广泛认可。然而,固有的辐射暴露、有限的时间和空间分辨率以及较差的敏感性促使人们寻求更安全和敏感的替代品。因此,迫切需要一种新的成像工具,能够以更高的分辨率和更精确的解剖特征实时跟踪体内循环系统的动态血流。对于以缺血性脑卒中为代表的治疗时间窗狭窄的血管性疾病,及时定位缺血脑区对药物干预具有重要意义。近红外二区荧光成像也需要大量的显影剂来满足各类的应用需求,近红外二区显像剂如单壁碳纳米管、量子点、稀土掺杂纳米颗粒、聚合物、小分子有机染料等,在生物医学研究中越来越广泛。其中,有机荧光小分子相比于无机的荧光团具有许多优点,如结构修饰方便、光谱易调控、容易代谢和生物相容性好等特点因而被视为更有潜力应用到临床上。
3.荧光成像已被证明是一种无辐射、高灵敏度和快速反馈的有价值的工具。事实上,在过去的几十年中,荧光成像主要位于可见光范围(400-650nm)到第一近红外(nir-i,650-950nm)的光谱窗口,只能达到几毫米以内的组织穿透深度,这在很大程度上限制了它们在临床实践中的应用。与可见光和nir-i光相比,第二近红外(nir-ii,1000-1700nm)光的荧光成像由于光子散射和组织自体荧光的显著减弱而产生无与伦比的时空分辨率和组织穿透,特别是第二个近红外子窗口(nir iib,1500
–
1700nm)可以实现较低的光子散射和最小的自荧光,受到越来越多的关注。
4.然而,广泛使用的无机nir-iib造影剂,如单壁碳纳米管、稀土掺杂下转换纳米颗粒和量子点,具有未知的长期毒性问题。因此,开发用于体内近红外iib成像的有机小分子荧光团迫在眉睫。先前已经报道了几种用于体内生物成像的有机nir iib造影剂。然而,有机荧光团的nir iib窗口中的量子产率(qy)仍然有限(《0.15%),这是由于荧光量子效率随着波长的延长和占主导地位的非辐射弛豫动力学而显著降低,尤其是在nir-ii荧光分子中。
5.为了获得具有优异性能的近红外二区荧光成像探针,研发一种具有高荧光强度、高的组织穿透性、光稳定性好、低毒性且具备nir-iib成像的新型小分子近红外二区荧光染料显得很有必要。
技术实现要素:
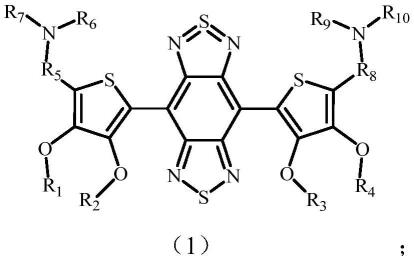
6.本发明旨在至少在一定程度上解决现有技术中存在的技术问题之一,由此,在本发明的第一方面,本发明提供一种1500nm以上成像的有机荧光小分子化合物,所述1500nm以上成像的有机荧光小分子化合物结构式如式(1)所示:
[0007][0008]
其中,r1、r2、r3、r4分别独自地选自c1-c12烷基、x取代的c1-c12烷基中的一种,;x选自f、cl、br、i、n3、cooh、oh、sh、cho、nh2、so3h、cn中的一种;当r1、r2同时为-ch
2-时,r1、r2以碳碳键相连;当r1、r2同时为-ch
2-时,r1、r2以碳碳键相连;
[0009]
r5、r8分别独自地选自中的一种,
[0010]
r6、r7、r9、r
10
分别独自地选自中的一种。
[0011]
优选地,r1、r2、r3、r4分别独自地选自c1-c10烷基。
[0012]
在本发明的一个或多个实施例中,所述1500nm以上成像的有机荧光小分子化合物选自如下化合物hy4、化合物hy3、化合物hy2、化合物hy1中的一种:
[0013]
[0014][0015]
优选地,所述1500nm以上成像的有机荧光小分子化合物选自化合物hy4、化合物hy3、化合物hy2中的一种;更优选地,所述1500nm以上成像的有机荧光小分子化合物为化合物hy4。
[0016]
在本发明的一个或多个实施例中,所述1500nm以上成像的有机荧光小分子化合物的荧光发射波长为1000~1700nm。
[0017]
在本发明的第二方面,本发明提供一种纳米颗粒,所述纳米颗粒包含本发明的第一方面所述的1500nm以上成像的有机荧光小分子化合物,优选地,所述纳米颗粒由二硬脂酰基磷脂酰乙醇胺-聚乙二醇与本发明的第一方面所述的1500nm以上成像的有机荧光小分子化合物自组装形成。
[0018]
在本发明的一个或多个实施例中,所述纳米颗粒的直径为20~200nm,优选为100~150nm。
[0019]
在本发明的第三方面,本发明提供一种本发明的第一方面所述的1500nm以上成像的有机荧光小分子化合物和/或本发明的第二方面所述的纳米颗粒在近红外二区1000~1700nm范围内的血管成像和血液类成像中的应用。
[0020]
在本发明的第四方面,本发明提供一种本发明的第一方面所述的1500nm以上成像的有机荧光小分子化合物的制备方法,所述化合物(1)由化合物(9)反应得到,所述化合物(9)制备化合物(1)的反应式如下所示:
[0021][0022]
化合物(9)制备化合物(1)包括如下步骤:将化合物(9)、化合物(10)加入反应容器中,在氮气或氩气保护下加入四氢呋喃使化合物(9)、化合物(10)溶解,随后向反应液中通入氮气或氩气排空氧气,再加入碳酸钾水溶液,并取[1,1'-双(二苯基膦)二茂铁]二氯化钯二氯甲烷络合物加入到反应容器中,继续向反应液中通入氮气或氩气,70~80℃条件下反应3~12小时,分离纯化,即得化合物(1);
[0023]
优选地,化合物(9)、化合物(10)、[1,1'-双(二苯基膦)二茂铁]二氯化钯二氯甲烷络合物和碳酸钾的摩尔比是1:(0.8~1.2):(0.005-0.05):(1.5~2.5);反应体系中,四氢
呋喃和水的体积比为(2~3):1。
[0024]
在本发明的一个或多个实施例中,所述化合物(9)由化合物(6)反应得到,所述化合物(6)制备化合物(9)的反应式如下所示:
[0025][0026]
化合物(6)制备化合物(9)包括如下步骤:
[0027]
步骤1):取化合物(6)、锌粉加入到反应容器中,在氮气或氩气保护下加入二氯甲烷和甲醇-水混合溶剂使其溶解,再加入氯化铵,室温反应1~3小时,反应结束后进行抽滤,取滤液进行萃取分液,有机相旋蒸去除溶剂得到第一中间产物;在氮气或氩气保护下,将第一中间产物加入到反应容器,随后加入吡啶使其溶解,取n-亚磺酰苯胺和三甲基氯硅烷加入到反应液中,在80~90℃条件下反应8~20小时,反应结束后进行分离纯化得到第二中间产物;在氮气或氩气保护下,将第二中间产物加入到反应容器中,加入n,n-二甲基甲酰胺-乙腈混合溶液使其溶解,将溴化氢滴加到反应溶液中,随后称取n-溴代丁二酰亚胺在反应过程中平均分三次加入,在60~70℃下避光反应3~6小时,分离纯化,即得化合物(7);
[0028]
步骤2):取化合物(7)、化合物(8)加入到反应容器中,在氮气或氩气保护下加入四氢呋喃使其溶解,随后向反应液中通入氮气或氩气排空氧气,再加入碳酸钾水溶液,并取[1,1'-双(二苯基膦)二茂铁]二氯化钯二氯甲烷络合物加入到反应容器中,继续向反应液中通入氮气或氩气,70~80℃条件下反应3~12小时,分离纯化,即得化合物(9);
[0029]
优选地,所述步骤1)中,化合物(6)、锌粉、氯化铵、n-亚磺酰苯胺、三甲基氯硅烷和n-溴代丁二酰亚胺的摩尔比为1:(50~70):(30~40):(50~70):(70~90):(2~2.5);所述的二氯甲烷、甲醇和水的体积比为(2~4):(1~3):1;所述的n,n-二甲基甲酰胺和乙腈的体积比为(1~3):1。
[0030]
更优选地,所述步骤2)中,化合物(7)、化合物(8)、[1,1'-双(二苯基膦)二茂铁]二氯化钯二氯甲烷络合物和碳酸钾的摩尔比是1:(0.8~1.2):(0.005-0.05):(1.5~2.5);反应体系中,四氢呋喃和水的体积比为(2~3):1。
[0031]
在本发明的一个或多个实施例中,所述化合物(6)由化合物(2)反应得到,所述化合物(2)制备化合物(6)的反应式如下所示:
[0032][0033]
化合物(2)制备化合物(6)包括如下步骤:
[0034]
步骤a:取化合物(2)、化合物(3)加入到反应容器中,在氮气或氩气保护下加入甲苯使其溶解;向反应液中通入氮气或氩气,排出系统内氧气,加入四(三苯基膦)钯,继续向反应液中通入氮气或氩气,加热回流反应4~24小时,分离纯化,即得化合物(4);
[0035]
步骤b:取化合物(4)、化合物(5)加入到反应容器中,在氮气或氩气保护下加入甲
苯使其溶解;向反应液中通入氮气或氩气,排出系统内氧气,加入四(三苯基膦)钯,继续向反应液中通入氮气或氩气,加热回流反应4~24小时,分离纯化,即得化合物(6);
[0036]
优选地,所述步骤a中,控制化合物(2)、化合物(3)和四(三苯基膦)钯的摩尔比为(0.8~1.2):1:(0.05~0.1);
[0037]
更优选地,所述步骤b中,控制化合物(5)、化合物(4)和四(三苯基膦)钯的摩尔比为(0.8~1.2):1:(0.05~0.1)。
[0038]
在本发明的第五方面,本发明提供一种在本发明的第一方面所述的1500nm以上成像的有机荧光小分子化合物和/或在本发明的第二方面所述的纳米颗粒在制备用于生物体内成像的近红外二区荧光成像探针中的用途。
[0039]
在本发明的第六方面,本发明提供一种近红外荧光成像探针,所述探针由本发明的第一方面所述的可调控代谢的有机荧光小分子化合物制备得到。
[0040]
本发明的有益效果在于:
[0041]
1、本发明提供一种可在1500nm以上荧光发射的有机荧光小分子化合物,其经二硬脂酰基磷脂酰乙醇胺-聚乙二醇包载后,可用于近红外二区血管,血液类疾病成像等;
[0042]
2、本发明提供一种可在1500nm以上荧光发射的有机荧光小分子化合物,其为具有最大发射波长超过1000nm的全新化合物,并且在1500nm以上具有较好的量子产率(qy=0.27%),无毒,生物相容性好,易被生物体吸收和代谢;
[0043]
3、本发明提供一种可调控代谢的有机荧光小分子化合物的制备方法,其合成路线简单,反应效率高,收率高,具有较高的工业应用前景;
[0044]
4、本发明提供一种近红外荧光成像探针,其由上述可在1500nm以上荧光发射的有机荧光小分子化合物制备得到,其在生物成像的实验中,可实现很好的时间和空间分辨率,具有很好的应用前景。
附图说明
[0045]
图1为化合物hy4核磁氢谱表征;
[0046]
图2为化合物hy3核磁碳谱表征;
[0047]
图3为化合物hy2核磁碳谱表征;
[0048]
图4为化合物hy1核磁碳谱表征;
[0049]
图5为化合物hy1、hy2、hy3和hy4在四氢呋喃中的吸收光谱图;
[0050]
图6为化合物hy1、hy2、hy3和hy4在四氢呋喃中的发射光谱图;
[0051]
图7化合物hy1、hy2、hy3和hy4的aie特性探究图;
[0052]
图8为化合物hy4纳米颗粒的透射电镜和动态光散射结果;
[0053]
图9为荧光探针hy1、hy2、hy3和hy4纳米颗粒在水中的吸收光谱图;
[0054]
图10为荧光探针hy1、hy2、hy3和hy4纳米颗粒在水中的发射光谱图;
[0055]
图11为荧光探针hy4纳米颗粒在不同滤光片(1000nm,1250nm,1320nm,1500nm)下的腿部血管成像图;
[0056]
图12为荧光探针hy4纳米颗粒在不同滤光片(1000nm,1250nm,1320nm,1500nm)下的腿部缺血成像图;
[0057]
图13为荧光探针hy4纳米颗粒在不同滤光片(1000nm,1250nm,1320nm,1500nm)下
的静脉和动脉血栓成像图;
[0058]
图14为荧光探针hy4纳米颗粒在不同滤光片(1000nm,1250nm,1320nm,1500nm)下的正常小鼠,脑梗小鼠,给药后的脑梗小鼠的脑部成像图以及荧光信号的分析;图14a为灯盏细辛注射液给药过程的示意图,图14b为注射荧光探针hy4纳米颗粒(dots)的正常小鼠(sham)、脑梗小鼠(vehicle)、给了灯盏细辛注射液后的脑梗小鼠(dxi)在不同滤光片(1000nm,1250nm,1320nm,1500nm)下近红外二区成像结果图,图14c为用高斯计算对血管的拟合的血管直径,图14d为通过荧光强度的方式分析药物的治疗效果图;
[0059]
图15为正常小鼠,脑梗小鼠,给了灯盏细辛注射液后的脑梗小鼠的ttc以及病理染色分析;
[0060]
图16左为正常小鼠(sham),脑梗小鼠(mcao),给了灯盏细辛注射液后的脑梗小鼠(dxi)的dapi染色图、tunel染色图和dapi、tune合并染色图(merged),图16右为正常小鼠(sham),脑梗小鼠(mcao),给了灯盏细辛注射液后的脑梗小鼠(dxi)的tunel阳性细胞比例对比图;
[0061]
图17左为正常小鼠(sham),脑梗小鼠(mcao),给了灯盏细辛注射液后的脑梗小鼠(dxi)的dapi染色图、cleaved caspase染色图和dapi、cleaved caspase合并染色图(merged),图16右为正常小鼠(sham),脑梗小鼠(mcao),给了灯盏细辛注射液后的脑梗小鼠(dxi)的tunel荧光强度对比图;
[0062]
图18左为正常小鼠(sham),脑梗小鼠(mcao),给了灯盏细辛注射液后的脑梗小鼠(dxi)的dapi染色图、gfap染色图、neun染色图和dapi、gfap、neun的合并染色图(merged),图18右为正常小鼠(sham),脑梗小鼠(mcao),给了灯盏细辛注射液后的脑梗小鼠(dxi)的tunel荧光强度对比图;
[0063]
图19左为正常小鼠(sham),脑梗小鼠(mcao),给了灯盏细辛注射液后的脑梗小鼠(dxi)的dapi染色图、cd31染色图、cd34染色图和dapi、cd31、cd34的合并染色图(merged),图19右为正常小鼠(sham),脑梗小鼠(mcao),给了灯盏细辛注射液后的脑梗小鼠(dxi)的tunel荧光强度对比图。
具体实施方式
[0064]
下面将结合实施例对本发明的方案进行解释。本领域技术人员将会理解,下面的实施例仅用于说明本发明,而不应视为限定本发明的范围。实施例中未注明具体技术或条件的,按照本领域内的文献所描述的技术或条件或者按照产品说明书进行。以下实施例中未注明具体条件者,按照常规条件或制造商建议的条件进行,使用的方法如无特别说明,均为本领域公知的常规方法,使用的耗材和试剂如无特别说明,均为市场购得。除非另有说明,本文中所用的专业与科学术语与本领域熟练人员所熟悉的意义相同。此外,任何与所记载内容相似或均等的方法或材料也可应用于本发明中。
[0065]
本发明提供一种1500nm以上成像的有机荧光小分子化合物,所述1500nm以上成像的有机荧光小分子化合物结构式如式1所示:
[0066][0067]
其中,r1、r2、r3、r4分别独自地选自c1-c12烷基、x取代的c1-c12烷基中的一种;x选自f、cl、br、i、n3、cooh、oh、sh、cho、nh2、so3h、cn中的一种;
[0068]
r5、r8分别独自地选自中的一种,
[0069]
r6、r7、r9、r
10
分别独自地选自中的一种。
[0070]
上述1500nm以上成像的有机荧光小分子化合物(式(1)所示化合物)的制备路线如下所示:
[0071][0072]
以下实施例1~4分别以hy4、hy3、hy2和hy1为示例具体化合物说明通式(1)的合成方法:
[0073]
实施例1:hy4的合成
[0074]
hy4的反应式如下所示:
[0075][0076]
化合物hy4的制备方法包括如下步骤:
[0077]
取化合物(2a)(1.5g,2.4mmol)、化合物(3)(737mg,1.9mmol)加入到反应容器中,在氮气或氩气保护下加入甲苯(30ml)使其溶解;向反应液中通入氮气或氩气,排出系统内氧气,加入四(三苯基膦)钯(219mg,0.19mmol),继续向反应液中通入氮气或氩气,加热回流反应15小时,反应结束后加入ea(20ml
×
3)萃取三次,合并有机相,水(10ml
×
2)洗两次。有机相加入适量无水硫酸钠干燥0.5h,过滤,滤液旋蒸后过硅胶柱进行分离纯化得到930mg中间体化合物(4a)。产率:76%。
[0078]
取化合物(4a)(930mg,1.4mmol)、化合物(5a)(1.1g,1.7mmol)加入到反应容器中,在氮气或氩气保护下加入甲苯(30ml)使其溶解;向反应液中通入氮气或氩气,排出系统内氧气,加入四(三苯基膦)钯(161mg,0.14mmol),继续向反应液中通入氮气或氩气,加热回流反应15小时,反应结束后加入乙酸乙酯(20ml
×
3)萃取三次,合并有机相,水(10ml
×
2)洗两次。有机相加入适量无水硫酸钠干燥0.5h,过滤,滤液旋蒸后过硅胶柱进行分离纯化得到1g中间体化合物(6a)。产率:79%。
[0079]
取中间体化合物(6a)(500mg,0.5mmol)、锌粉(2g,33mmol)加入到反应容器中,在氮气或氩气保护下加入二氯甲烷(15ml)和甲醇-水混合溶剂(v/v=2:1,7.5ml)使其溶解,再加入氯化铵(1g,19.8mmol),室温反应2h,反应结束后使用硅藻土抽滤,二氯甲烷(10ml
×
2)洗滤饼两次,取滤液加入二氯甲烷(15ml
×
3)萃取三次,水(10ml
×
2)洗两次,有机相加入适量无水硫酸钠干燥0.5h,过滤后取滤液旋蒸得到第一中间产物;在氮气或氩气保护下,将第一中间产物加入到反应容器,随后加入吡啶(8ml)使其溶解,取n-亚磺酰苯胺(4ml,35.5mmol)和三甲基氯硅烷(4ml,46.1mmol)加入到反应液中,在85℃条件下反应12h,反应结束后将反应液加入到冰水(20ml)中,再加入二氯甲烷(10ml
×
2)、1m稀盐酸(20ml
×
2)萃
取两次,取有机相加入适量无水硫酸钠干燥0.5h,过滤后取滤液进行旋蒸并过硅胶柱得到第二中间产物,;在氮气或氩气保护下,将第二中间产物加入到反应容器中,加入n,n-二甲基甲酰胺-乙腈混合溶液(v/v=2:1,27ml)使其溶解,滴加2滴溴化氢到反应溶液中,随后称取n-溴代丁二酰亚胺(53mg,0.3mmol)在反应过程中平均分三次加入,在65℃下避光反应3h,反应结束后加入二氯甲烷(15ml
×
3)萃取三次,水(10ml
×
2)洗两次,取有机相加入适量无水硫酸钠干燥0.5h,然后过滤并取滤液进行旋蒸、过硅胶柱分离纯化得到302mg中间体化合物(7a)。产率:50%。
[0080]
取中间体化合物(7a)(100mg,0.1mmol)、化合物(8)(42mg,0.1mmol)加入到反应容器中,在氮气或氩气保护下加入四氢呋喃(7ml)使其溶解,随后向反应液中通入氮气或氩气排空氧气,再加入碳酸钾水溶液(15mg/ml,2ml),并取[1,1'-双(二苯基膦)二茂铁]二氯化钯二氯甲烷络合物(8mg,0.01mmol)加入到反应中,继续向反应液中通入氮气或氩气,75℃条件下反应5小时,反应结束后加入二氯甲烷(10ml
×
3)萃取三次,水(5ml
×
2)洗两次,合并有机相,加入适量无水硫酸钠干燥0.5h,随后过滤并取滤液进行旋蒸并过硅胶柱进行分离纯化得到66mg中间体化合物(9a)。产率:55%。
[0081]
取中间体化合物(9a)(66mg,0.05mmol)、化合物(8)(21mg,0.05mmol)加入到反应容器中,在氮气或氩气保护下加入四氢呋喃(7ml)使其溶解,随后向反应液中通入氮气或氩气排空氧气,再加入碳酸钾水溶液(15mg/ml,2ml),并取[1,1'-双(二苯基膦)二茂铁]二氯化钯二氯甲烷络合物(4mg,0.005mmol)加入到反应中,继续向反应液中通入氮气或氩气,75℃条件下反应5小时,反应结束后加入二氯甲烷(10ml
×
3)萃取三次,水(5ml
×
2)洗两次,合并有机相,加入适量无水硫酸钠干燥0.5h,随后过滤并取滤液进行旋蒸并过硅胶柱进行分离纯化得到35mg化合物hy4。产率:48%。化合物hy4核磁氢谱表征如图1所示,1h nmr(400mhz,cdcl3)δ7.95(dd,j=18.4,8.3hz,4h),7.83(d,j=8.2hz,2h),7.67(d,j=8.7hz,4h),7.51(m,4h),7.40(m,4h),7.27(dd,j=13.2,4.8hz,4h),7.14(d,j=7.7hz,4h),7.05(d,j=8.8hz,8h),4.03(d,j=6.0hz,4h),3.87(t,j=5.6hz,4h),1.71(d,j=6.1hz,4h),1.52(dd,j=14.2,6.9hz,8h),1.37(m,12h),1.01(ddd,j=15.4,10.9,5.7hz,12h),0.88(m,12h),0.74(t,j=7.0hz,6h),0.55(t,j=7.4hz,6h).
13
c nmr(101mhz,cdcl3)δ152.77,150.74,148.03,147.92,145.81,143.25,135.32,131.20,129.52,129.20,128.44,128.17,127.29,126.66,126.47,126.37,126.20,126.09,124.25,122.50,122.22,121.00,114.28,114.17,110.00,76.17,75.70,53.45,40.29,40.11,30.33,30.14,29.10,28.97,23.59,23.34,23.10,22.96,14.15,14.04,11.14,10.83.maldi-tof-ms calcd for:c
90h100
n6o4s4([m+h]+):1456.67,found:1457.49.
[0082]
实施例2:hy3的合成
[0083]
hy3的反应式如下所示:
[0084][0085]
化合物hy2的制备方法包括如下步骤:
[0086]
取化合物(2b)(688mg,1.2mmol)、化合物(3)(384mg,1mmol)加入到反应容器中,在氮气或氩气保护下加入甲苯(20ml)使其溶解;向反应液中通入氮气或氩气,排出系统内氧气,加入四(三苯基膦)钯(115mg,0.1mmol),继续向反应液中通入氮气或氩气,加热回流反应15小时,反应结束后加入ea(15ml
×
3)萃取三次,合并有机相,水(8ml
×
2)洗两次。有机相加入适量无水硫酸钠干燥0.5h,过滤,滤液旋蒸后过硅胶柱进行分离纯化得到564mg中间体化合物(4b)。产率:80%。
[0087]
取化合物(4b)(564mg,0.96mmol)、化合物(5b)(307mg,0.8mmol)加入到反应容器中,在氮气或氩气保护下加入甲苯(20ml)使其溶解;向反应液中通入氮气或氩气,排出系统内氧气,加入四(三苯基膦)钯(92mg,0.08mmol),继续向反应液中通入氮气或氩气,加热回流反应15小时,反应结束后加入乙酸乙酯(15ml
×
3)萃取三次,合并有机相,水(8ml
×
2)洗两次。有机相加入适量无水硫酸钠干燥0.5h,过滤,滤液旋蒸后过硅胶柱进行分离纯化得到570mg中间体化合物(6b)。产率:75%。
[0088]
取中间体化合物(6b)(570mg,0.7mmol)、锌粉(3g,46mmol)加入到反应容器中,在氮气或氩气保护下加入二氯甲烷(20ml)和甲醇-水混合溶剂(v/v=2:1,10ml)使其溶解,再加入氯化铵(1.5g,28mmol),室温反应2h,反应结束后使用硅藻土抽滤,二氯甲烷(10ml
×
2)洗滤饼两次,取滤液加入二氯甲烷(15ml
×
3)萃取三次,水(10ml
×
2)洗两次,有机相加入适量无水硫酸钠干燥0.5h,过滤后取滤液旋蒸得到第一中间产物;在氮气或氩气保护下,将第一中间产物加入到反应容器,随后加入吡啶(11ml)使其溶解,取n-亚磺酰苯胺(5.6ml,49.7mmol)和三甲基氯硅烷(5.6ml,64.5mmol)加入到反应液中,在85℃条件下反应12h,反应结束后将反应液加入到冰水(25ml)中,再加入二氯甲烷(15ml
×
2)、1m稀盐酸(28ml
×
2)萃取两次,取有机相加入适量无水硫酸钠干燥0.5h,过滤后取滤液进行旋蒸并过硅胶柱得到第二中间产物,在氮气或氩气保护下,将第二中间产物加入到反应容器中,加入n,n-二甲基甲酰胺-乙腈混合溶液(v/v=2:1,38ml)使其溶解,滴加4滴溴化氢到反应溶液中,随后称取n-溴代丁二酰亚胺(74.8mg,0.42mmol)在反应过程中平均分三次加入,在65℃下避光反应3h,反应结束后加入二氯甲烷(20ml
×
3)萃取三次,水(14ml
×
2)洗两次,取有机相加入适量无水硫酸钠干燥0.5h,然后过滤并取滤液进行旋蒸、过硅胶柱分离纯化得到353mg中间体化合物(7b)。产率:55%。
[0089]
取中间体化合物(7b)(100mg,0.11mmol)、化合物(8)(46mg,0.11mmol)加入到反应
容器中,在氮气或氩气保护下加入四氢呋喃(7ml)使其溶解,随后向反应液中通入氮气或氩气排空氧气,再加入碳酸钾溶液(15mg/ml,2ml),并取[1,1'-双(二苯基膦)二茂铁]二氯化钯二氯甲烷络合物(8mg,0.01mmol)加入到反应中,继续向反应液中通入氮气或氩气,75℃条件下反应5小时,反应结束后加入二氯甲烷(10ml
×
3)萃取三次,水(5ml
×
2)洗两次,合并有机相,加入适量无水硫酸钠干燥0.5h,随后过滤并取滤液进行旋蒸并过硅胶柱进行分离纯化得到72mg中间体化合物(9b)。产率:58%。
[0090]
取中间体化合物(9b)(72mg,0.06mmol)、化合物(8)(25mg,0.06mmol)加入到反应容器中,在氮气或氩气保护下加入四氢呋喃(7ml)使其溶解,随后向反应液中通入氮气或氩气排空氧气,再加入碳酸钾水溶液(15mg/ml,2ml),并取[1,1'-双(二苯基膦)二茂铁]二氯化钯二氯甲烷络合物(4mg,0.005mmol)加入到反应中,继续向反应液中通入氮气或氩气,75℃条件下反应5小时,反应结束后加入二氯甲烷(10ml
×
3)萃取三次,水(5ml
×
2)洗两次,合并有机相,加入适量无水硫酸钠干燥0.5h,随后过滤并取滤液进行旋蒸并过硅胶柱进行分离纯化得到42mg化合物hy3。产率:52%。化合物hy3核磁氢谱表征如图2所示,1h nmr(400mhz,cdcl3)δ7.95(dd,j=17.3,8.3hz,4h),7.83(d,j=8.2hz,2h),7.70(d,j=8.8hz,4h),7.51(dt,j=11.0,7.8hz,4h),7.40(m,4h),7.26(m,4h),7.15(d,j=7.6hz,4h),7.03(m,8h),4.12(q,j=6.8hz,8h),1.77(m,4h),1.43(dd,j=13.9,7.1hz,8h),1.32(m,12h),1.09(m,8h),0.90(dd,j=9.1,4.6hz,6h),0.78(t,j=7.0hz,6h).
13
c nmr(101mhz,cdcl3)δ152.78,150.37,149.05,147.93,145.65,143.19,139.73,135.32,131.60,131.22,129.81,129.21,128.44,127.79,127.35,126.70,126.49,126.38,126.22,126.00,124.24,122.58,122.29,121.96,120.91,114.38,114.16,110.17,110.00,73.80,73.42,72.96,72.89,31.65,31.52,31.46,31.18,30.18,30.11,30.01,27.56,25.74,25.60,25.47,25.13,24.86,23.68,22.63,22.49,20.13,14.08,13.98.maldi-tof-ms calcd for:c
82h84
n6o4s4([m+h]+):1344.54,found:1345.22.
[0091]
实施例3:hy2的合成
[0092]
hy2的反应式如下所示:
[0093][0094]
化合物hy2的制备方法包括如下步骤:
[0095]
取化合物(2c)(1g,2.3mmol)、化合物3(845mg,2.2mmol)加入到反应容器中,在氮气或氩气保护下加入甲苯(30ml)使其溶解;向反应液中通入氮气或氩气,排出系统内氧气,加入四(三苯基膦)钯(254mg,0.22mmol),继续向反应液中通入氮气或氩气,加热回流反应15小时,反应结束后加入ea(20ml
×
3)萃取三次,合并有机相,水(10ml
×
2)洗两次。有机相加入适量无水硫酸钠干燥0.5h,过滤,滤液旋蒸后过硅胶柱进行分离纯化得到738mg中间体
化合物(4c)。产率:75%。
[0096]
取化合物(4c)(738mg,1.65mmol)、化合物(5c)(736mg,1.7mmol)加入到反应容器中,在氮气或氩气保护下加入甲苯(30ml)使其溶解;向反应液中通入氮气或氩气,排出系统内氧气,加入四(三苯基膦)钯(185mg,0.16mmol),继续向反应液中通入氮气或氩气,加热回流反应15小时,反应结束后加入乙酸乙酯(20ml
×
3)萃取三次,合并有机相,水(10ml
×
2)洗两次。有机相加入适量无水硫酸钠干燥0.5h,过滤,滤液旋蒸后过硅胶柱进行分离纯化得到589mg中间体化合物(6c)。产率:70%。
[0097]
取中间体化合物(6c)(589mg,1.2mmol)、锌粉(5.1g,78mmol)加入到反应容器中,在氮气或氩气保护下加入二氯甲烷(35ml)和甲醇-水混合溶剂(v/v=2:1,15ml)使其溶解,再加入氯化铵(2.5g,47.6mmol),室温反应2h,反应结束后使用硅藻土抽滤,二氯甲烷(20ml
×
2)洗滤饼两次,取滤液加入二氯甲烷(25ml
×
3)萃取三次,水(15ml
×
2)洗两次,有机相加入适量无水硫酸钠干燥0.5h,过滤后取滤液旋蒸得到第一中间产物;在氮气或氩气保护下,将第一中间产物加入到反应容器,随后加入吡啶(19ml)使其溶解,取n-亚磺酰苯胺(9.5ml,84.5mmol)和三甲基氯硅烷(9.5ml,109.6mmol)加入到反应液中,在85℃条件下反应12h,反应结束后将反应液加入到冰水(35ml)中,再加入二氯甲烷(25ml
×
2)、1m稀盐酸(35ml
×
2)萃取两次,取有机相加入适量无水硫酸钠干燥0.5h,过滤后取滤液进行旋蒸并过硅胶柱得到第二中间产物,;在氮气或氩气保护下,将第二中间产物加入到反应容器中,加入n,n-二甲基甲酰胺-乙腈混合溶液(v/v=2:1,65ml)使其溶解,滴加6滴溴化氢到反应溶液中,随后称取n-溴代丁二酰亚胺(126mg,0.71mmol)在反应过程中平均分三次加入,在65℃下避光反应3h,反应结束后加入二氯甲烷(30ml
×
3)萃取三次,水(20ml
×
2)洗两次,取有机相加入适量无水硫酸钠干燥0.5h,然后过滤并取滤液进行旋蒸、过硅胶柱分离纯化得到366mg中间体化合物(7c)。产率:48%。
[0098]
取中间体化合物(7c)(80mg,0.13mmol)、化合物(8)(63mg,0.15mmol)加入到反应容器中,在氮气或氩气保护下加入四氢呋喃(10ml)使其溶解,随后向反应液中通入氮气或氩气排空氧气,再加入碳酸钾水溶液(15mg/ml,2.2ml),并取[1,1'-双(二苯基膦)二茂铁]二氯化钯二氯甲烷络合物(8mg,0.01mmol)加入到反应中,继续向反应液中通入氮气或氩气,75℃条件下反应5小时,反应结束后加入二氯甲烷(12ml
×
3)萃取三次,水(6ml
×
2)洗两次,合并有机相,加入适量无水硫酸钠干燥0.5h,随后过滤并取滤液进行旋蒸并过硅胶柱进行分离纯化得到50mg中间体化合物(9c)。产率:45%。
[0099]
取中间体化合物(9c)(50mg,0.06mmol)、化合物(8)(29mg,0.07mmol)加入到反应容器中,在氮气或氩气保护下加入四氢呋喃(10ml)使其溶解,随后向反应液中通入氮气或氩气排空氧气,再加入碳酸钾水溶液(15mg/ml,2.2ml),并取[1,1'-双(二苯基膦)二茂铁]二氯化钯二氯甲烷络合物(4.9mg,0.006mmol)加入到反应中,继续向反应液中通入氮气或氩气,75℃条件下反应5小时,反应结束后加入二氯甲烷(12ml
×
3)萃取三次,水(6ml
×
2)洗两次,合并有机相,加入适量无水硫酸钠干燥0.5h,随后过滤并取滤液进行旋蒸并过硅胶柱进行分离纯化得到27mg化合物hy2。产率:43%。化合物hy2核磁氢谱表征如图3所示,1h nmr(400mhz,cdcl3)δ7.95(dd,j=15.3,8.3hz,4h),7.83(d,j=8.2hz,2h),7.67(d,j=8.7hz,4h),7.52(m,4h),7.40(dd,j=12.0,6.5hz,4h),7.27(m,4h),7.16(d,j=7.9hz,4h),7.03(t,j=7.8hz,8h),3.95(d,j=7.8hz,8h).maldi-tof-ms calcd for:c
62h44
n6o4s4([m+h]
+):1064.23,found:1063.89.
[0100]
实施例4
[0101]
用实施例1、2、3同样的方法以化合物(2d)替代化合物(2a)或化合物(2b)或化合物(2c)制备hy1。化合物(2d)的结构如下所示:
[0102][0103]
化合物hy1的结构如下所示:
[0104][0105]
化合物hy1核磁氢谱表征如图4所示。
[0106]
图5为化合物hy1、hy2、hy3、hy4在四氢呋喃的吸收光谱图;图6为化合物hy1、hy2、hy3、hy4在四氢呋喃的发射光谱图;图7化合物hy1、hy2、hy3、hy4的aie特性探究图;由图可知,hy1不具有aie特性,hy2、hy3、hy4均具有显著的aie特性,并且hy4的aie特性最强。
[0107]
实施例5:荧光探针hy1、hy2、hy3、hy4纳米颗粒的制备
[0108]
实施例1制备得到的化合物hy4、实施例2制备得到的化合物hy3、实施例3制备得到的化合物hy2、实施例4制备得到的化合物hy1分别转变为可用于生物成像探针hy4、hy3、hy2、hy1纳米颗粒的制备具体步骤如下所示:
[0109]
以dspe-peg3k(二硬脂酰基磷脂酰乙醇胺-聚乙二醇)为包封基质,分别以实施例1制备得到的hy4、实施例2制备得到的hy3、实施例3制备得到的hy2、实施例4制备得到的hy1为原料采用纳米沉淀法对应制备得到hy4纳米颗粒、hy3纳米颗粒、hy2纳米颗粒、hy1纳米颗粒。随后,在冰浴中恒定超声作用下,分别将得到的hy4纳米颗粒、hy3纳米颗粒、hy2纳米颗粒、hy1纳米颗粒放入四氢呋喃中(纳米颗粒的浓度均为1mg/ml),分别将得到的四种纳米颗粒的四氢呋喃混合液逐滴添加到含有dspe-peg3k(二硬脂酰基磷脂酰乙醇胺-聚乙二醇)10mg的10ml去离子水溶液中。然后,在惰性气流下完全去除混合物中残留的四氢呋喃。利用40kda离心过滤工具,通过超滤离心去除多余的dspe-peg3k(二硬脂酰基磷脂酰乙醇胺-聚乙二醇),得到荧光探针hy4纳米颗粒、荧光探针hy3纳米颗粒、荧光探针hy2纳米颗粒、荧光探针hy1纳米颗粒颗粒。
[0110]
图8为化合物hy4纳米颗粒的透射电镜和动态光散射结果;由结果可知,hy4纳米颗粒的粒径为100~150nm,平均水和粒径为100~150nm;图9为荧光探针hy1-hy4纳米颗粒颗
粒在水中的吸收光谱图;图10为荧光探针hy1、hy2、hy3、hy4纳米颗粒颗粒在水中的发射光谱图;由图可知,荧光探针hy1、hy2、hy3、hy4纳米颗粒可以进一步用于近红外二区生物成像。
[0111]
实施例6
[0112]
以下实验为实施例5得到的荧光探针hy4纳米颗粒(dots)对小鼠的缺血腿部血管成像荧光探针hy4纳米颗粒(dots)对小鼠的缺血腿部血管成像。具体步骤如下:
[0113]
使用戊巴比妥钠将小鼠麻醉,通过尾静脉注射200微升的荧光探针hy4纳米颗粒(dots),使用功率密度110mw/cm2的808nm激光器照射小鼠。相机镜头前分别加1000nm、1250nm、1320nm、1500nm的长通滤光片。在注射5min后对小鼠腿部血管的荧光图像,结果如图11所示,由图可知,荧光探针hy4纳米颗粒(dots)在通过尾静脉注射到正常小鼠体内后,可以清晰的观察到小鼠小腿处的血管,尤其是1500nm下的血管具有更好的成像分辨率。
[0114]
实施例7
[0115]
以下实验为实施例5得到的荧光探针hy4纳米颗粒(dots)对小鼠腿部缺血部位的成像荧光探针hy4纳米颗粒(dots)对小鼠腿部缺血部位的成像。具体步骤如下:
[0116]
使用戊巴比妥钠将小鼠麻醉,通过尾静脉注射200微升的荧光探针hy4纳米颗粒(dots),使用功率密度110mw/cm2的808nm激光器照射小鼠。相机镜头前分别加1000nm、1250nm、1320nm、1500nm的长通滤光片。在注射5min后对小鼠腿部血管的荧光图像,结果如图12所示,由图可知,荧光探针hy4纳米颗粒(dots)在通过尾静脉注射到正常小鼠体内后,可以清晰的观察到小鼠小腿处缺血部位的血管图,尤其是1500nm下的缺血部位具有更好的成像分辨率。
[0117]
实施例8
[0118]
以下实验为实施例5得到的荧光探针hy4纳米颗粒(dots)对小鼠的血栓成像
[0119]
荧光探针hy4纳米颗粒(dots)对小鼠的血栓成像。具体步骤如下:
[0120]
使用戊巴比妥钠将小鼠麻醉,通过尾静脉注射200微升荧光探针hy4纳米颗粒(dots),使用功率密度110mw/cm2的808nm激光器照射小鼠。相机镜头前分别加1000nm、1250nm、1320nm、1500nm的长通滤光片。对小鼠全身的血管收集注射5min后的荧光图像,图13依次为荧光探针hy4纳米颗粒在1000nm,1250nm,1320nm,1500nm滤光片下的静脉和动脉血栓成像图;由图可知,荧光探针hy4纳米颗粒(dots)在通过尾静脉注射到患有血栓小鼠体内后,可以清晰的观察到小鼠腹部处的血栓和腿部的血栓,尤其是1500nm下的血栓处具有更好的成像分辨率。
[0121]
实施例9
[0122]
以下实验为实施例5得到的荧光探针hy4纳米颗粒(dots)对小鼠的脑梗成像
[0123]
荧光探针hy4纳米颗粒(dots)对小鼠的脑梗成像。具体步骤如下:
[0124]
使用戊巴比妥钠将小鼠麻醉,通过尾静脉注射200微升荧光探针hy4纳米颗粒(dots),使用功率密度100mw/cm2的808nm激光器照射正常小鼠(sham),脑梗小鼠(vehicle),给了灯盏细辛注射液后的脑梗小鼠(dxi)。相机镜头前分别加1000nm、1250nm、1320nm、1500nm的长通滤光片。对小鼠脑部的血管收集注射5min后的荧光图像,结果如图14所示,图14a为灯盏细辛注射液给药过程的示意图,图14b为注射荧光探针hy4纳米颗粒(dots)的正常小鼠(sham)、脑梗小鼠(vehicle)、给了灯盏细辛注射液后的脑梗小鼠(dxi)
在不同滤光片(1000nm,1250nm,1320nm,1500nm)下近红外二区成像结果图,图14c为用高斯计算对血管的拟合的血管直径,图14d为通过荧光强度的方式分析药物的治疗效果图,由图可知,探针hy4 dots在通过尾静脉注射到正常小鼠、脑梗小鼠和给药(灯盏细辛注射液)后的脑梗小鼠体内后,可以清晰的观察到小鼠的脑部血管的差异,在给了灯盏细辛注射液后的灯盏细辛注射液对小鼠的脑部有一定的保护作用,尤其是1500nm下的血栓处具有更好的成像分辨率。
[0125]
图15为正常小鼠(sham),脑梗小鼠(vehicle),给了灯盏细辛注射液后的脑梗小鼠(dxi)的ttc以及病理染色结果图;
[0126]
图16左为正常小鼠(sham),脑梗小鼠(mcao),给了灯盏细辛注射液后的脑梗小鼠(dxi)的dapi染色图、tunel染色图和dapi、tune合并染色图(merged),图16右为正常小鼠(sham),脑梗小鼠(mcao),给了灯盏细辛注射液后的脑梗小鼠(dxi)的tunel阳性细胞比例对比图,图16结果表明,mcao小鼠tunel阳性细胞比例较假手术组明显升高,而给了灯盏细辛注射液(dxi)后的脑梗小鼠可部分消除升高趋势;
[0127]
图17左为正常小鼠(sham),脑梗小鼠(mcao),给了灯盏细辛注射液后的脑梗小鼠(dxi)的dapi染色图、cleaved caspase染色图和dapi、cleaved caspase合并染色图(merged),图16右为正常小鼠(sham),脑梗小鼠(mcao),给了灯盏细辛注射液后的脑梗小鼠(dxi)的tunel荧光强度对比图,图17结果表明,mcao小鼠tunel荧光强度较假手术组明显升高,而给了灯盏细辛注射液(dxi)后的脑梗小鼠可部分消除升高趋势;
[0128]
图18左为正常小鼠(sham),脑梗小鼠(mcao),给了灯盏细辛注射液后的脑梗小鼠(dxi)的dapi染色图、gfap染色图、neun染色图和dapi、gfap、neun的合并染色图(merged),图18右为正常小鼠(sham),脑梗小鼠(mcao),给了灯盏细辛注射液后的脑梗小鼠(dxi)的tunel荧光强度对比图,图18结果表明,mcao小鼠tunel荧光强度较假手术组明显升高,而给了灯盏细辛注射液(dxi)后的脑梗小鼠可部分消除升高趋势;
[0129]
图19左为正常小鼠(sham),脑梗小鼠(mcao),给了灯盏细辛注射液后的脑梗小鼠(dxi)的dapi染色图、cd31染色图、cd34染色图和dapi、cd31、cd34的合并染色图(merged),图19右为正常小鼠(sham),脑梗小鼠(mcao),给了灯盏细辛注射液后的脑梗小鼠(dxi)的tunel荧光强度对比图,图19结果表明,mcao小鼠tunel荧光强度较假手术组明显升高,而给了灯盏细辛注射液(dxi)后的脑梗小鼠可部分消除升高趋势;
[0130]
图15~19结果表明,dxi通过促进缺血性脑卒中后的血管生成而减轻脑缺血损伤,因此,在nir-iib成像中,mcao小鼠tunel阳性细胞比例、tunel荧光强度较假手术组明显升高,而给了灯盏细辛注射液(dxi)后的脑梗小鼠可部分消除升高趋势,本发明荧光探针能清晰地观测到这一过程。
[0131]
尽管上面已经示出和描述了本发明的实施例,可以理解的是,上述实施例是示例性的,不能理解为对本发明的限制,本领域的普通技术人员在本发明的范围内可以对上述实施例进行变化、修改、替换和变型。
- 该技术已申请专利。仅供学习研究,如用于商业用途,请联系技术所有人。
- 技术研发人员:肖玉玲 李扬 洪学传 崔岩
- 技术所有人:武汉大学深圳研究院
- 我是此专利的发明人
- 该领域下的技术专家
- 如您需求助技术专家,请点此查看客服电话进行咨询。
- 1、薛老师:1.CRISPR-Cas系统 2.基因编辑 3.基因修复 4.天然产物合成 5.单分子技术开发与应用
- 2、张老师:1.探索新型氧化还原酶结构-功能关系,电催化反应机制 2.酶电催化导向的酶分子改造 3.纳米材料、生物功能多肽对酶-电极体系的影响4. 生物电化学传感和生物电合成体系的设计与应用。
- 3、豆老师:1.环境纳米材料及挥发性有机化合物(VOCs) 2.CO污染物的催化氧化 3.低温等离子体 4.吸脱附等控制技术
- 4、赵老师:1.高分子材料改性及加工技术 2.微孔及过滤材料 3.环境友好高分子材料
- 5、邬老师:1.高分子材料的共混与复合 2.涉及材料功能化及结构与性能的研究; 高分子热稳定剂的研发
- 如您是高校老师,可以点此联系我们加入专家库。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1
精彩留言,会给你点赞!
专利分类正在加载中....