一种2-氯-4-硝基苯-α-L-岩藻糖苷的制备方法与流程
一种2
‑
氯
‑4‑
硝基苯
‑
α
‑
l
‑
岩藻糖苷的制备方法
技术领域
1.本发明涉及一种2
‑
氯
‑4‑
硝基苯
‑
α
‑
l
‑
岩藻糖苷的制备方法,属于体外诊断领域。
背景技术:
2.α
‑
l
‑
岩藻糖苷酶(afu)是一种溶酶体酸性水解酶,广泛分布于人体内的各个组织、细胞及体液中。以肝、肾等组织活性较高,基本功能是催化含岩藻糖基的低聚糖、糖蛋白及糖苷的水解代谢,其本质是一种糖蛋白,在包括免疫反应、信号转导等多种方面发挥重要作用。研究发现,α
‑
l
‑
岩藻糖苷酶的测定在多种疾病的诊断及治疗中都有积极的作用。其中,速率法(连续监测法)血清中afu催化2
‑
氯
‑
对硝基酚α
‑
l
‑
岩藻糖苷(cnp
‑
afu)水解生成2
‑
氯
‑4‑
硝基苯酚(cnp),在405nm或410nm波长监测cnp的生成速率,计算出afu活性。本法测定对胆红素250mg/l、血红蛋白230mg/l、抗坏血酸6g/l无明显干扰。该法操作简便,测定时间短,灵敏度和抗干扰性有一定程度的提高。
3.目前,2
‑
氯
‑4‑
硝基苯
‑
α
‑
l
‑
岩藻糖苷的市场需求量较大,但是合成上有较大难度。该产品的合成方法包括:1)koichi等在1994年发表了一篇日本专利(jp 06
‑
179690),首次公开了2
‑
氯
‑4‑
硝基苯
‑
α
‑
l
‑
岩藻糖苷的合成方法;但该方法重现性较差,且需要用到大量的2
‑
氯
‑4‑
硝基苯酚,反应后处理十分困难。2)杜宇国等在2004年报道了一种新的合成路线;但该方法的起始原料乙基
‑1‑
巯
‑
β
‑
l
‑
岩藻糖苷和保护基原料叔丁基二甲基氯硅烷的价格昂贵,且反应收率较低,不适合工业化生产。3)2013年,北京利德曼生化股份有限公司也报道了一种新的合成路线;该方法得到的α构型产物比例高,但合成路线步骤较长,且需要用到乙硫醇,臭味大。因此,亟需寻找一种简便、实用的制备2
‑
氯
‑4‑
硝基苯
‑
α
‑
l
‑
岩藻糖苷的方法。
技术实现要素:
4.[技术问题]
[0005]
现有的制备方法的重现性差、后处理困难,或者成本高、收率低或合成步骤繁琐、使用臭味大的乙硫醇等物质。
[0006]
[技术方案]
[0007]
为了解决上述问题,本发明提供了一种新的2
‑
氯
‑4‑
硝基苯
‑
α
‑
l
‑
岩藻糖苷制备方法。本发明方法反应路线较短,通过三步反应制得最终产品;且以l
‑
岩藻糖为原料,起始原料价格相对便宜,2
‑
氯
‑4‑
硝基苯酚用量明显减少;后处理过程简单且反应重现性好,可实现工业化生产。
[0008]
具体的,本发明提供了一种2
‑
氯
‑4‑
硝基苯
‑
α
‑
l
‑
岩藻糖苷的制备方法,所述方法的反应路线如下:
[0009][0010]
所述方法包括以下步骤:
[0011]
(1)以l
‑
岩藻糖为起始原料,通过乙酰化反应,制得1,2,3,4
‑
四乙酰基
‑
α
‑
l
‑
岩藻糖;
[0012]
(2)1,2,3,4
‑
四乙酰基
‑
α
‑
l
‑
岩藻糖与2
‑
氯
‑4‑
硝基苯酚发生糖苷化反应,制得2
‑
氯
‑4‑
硝基苯
‑
2,3,4
‑
三乙酰基
‑
α
‑
l
‑
岩藻糖苷,其中,反应溶剂为二氯甲烷,三乙胺作为缚酸剂,所述催化剂选择三氟甲磺酸三甲基硅酯(tmsotf)或三氟化硼乙醚(bf3·
et2o)中的一种;
[0013]
(3)2
‑
氯
‑4‑
硝基苯
‑
2,3,4
‑
三乙酰基
‑
α
‑
l
‑
岩藻糖苷进行脱乙酰基反应,制得2
‑
氯
‑4‑
硝基苯
‑
α
‑
l
‑
岩藻糖苷。
[0014]
在本发明的一种实施方式中,步骤(1)中,所述乙酰化反应以碱作为催化剂,其中碱选自吡啶、醋酸钠中的一种,优选醋酸钠;乙酰化试剂选自乙酸酐、乙酸和乙酰氯中的一种或几种,优选乙酸酐。
[0015]
在本发明的一种实施方式中,步骤(1)中,所述催化剂的用量为1
‑
1.2当量,所述乙酰化试剂的用量为4
‑
5当量(每一步的用量,无特殊说明,默认为该步反应的起始原料)。
[0016]
在本发明的一种实施方式中,步骤(1)中,所述乙酰化反应的反应温度为25
‑
120℃,优选60
‑
100℃,反应时间为4
‑
12h。
[0017]
在本发明的一种实施方式中,步骤(1)中,乙酰化反应结束后,加入二氯甲烷中,水洗多次后经饱和氯化钠溶液清洗,之后干燥、除去溶剂即可得到1,2,3,4
‑
四乙酰基
‑
α
‑
l
‑
岩藻糖。
[0018]
在本发明的一种实施方式中,步骤(2)中,所述催化剂优选三氟化硼乙醚;催化剂用量4
‑
10当量,优选5
‑
8当量。
[0019]
在本发明的一种实施方式中,步骤(2)中,所述2
‑
氯
‑4‑
硝基苯酚用量1
‑
4当量,优选1.5
‑
2.5当量。
[0020]
在本发明的一种实施方式中,步骤(2)中,所述糖苷化反应的反应温度25
‑
50℃,优选40
‑
50℃,反应时间为48
‑
72h。
[0021]
在本发明的一种实施方式中,步骤(2)的反应在惰性气氛下进行,所述惰性气氛为氮气或氩气。
[0022]
在本发明的一种实施方式中,步骤(2)中,反应后处理采用重结晶方法获得2
‑
氯
‑4‑
硝基苯
‑
2,3,4
‑
三乙酰基
‑
α
‑
l
‑
岩藻糖苷,重结晶溶剂选自甲醇、乙醇、异丙醇中的一种或几种,避免了柱层析,大大降低了后处理难度。
[0023]
在本发明的一种实施方式中,步骤(3)中,所述脱乙酰基反应以碱作为催化剂,其中碱选自甲醇钠、乙醇钠、叔丁醇钠、叔丁醇钾、碳酸钠、碳酸钾、氢氧化钠、氢氧化钾、氢氧化锂中的一种或几种,优选甲醇钠、乙醇钠、叔丁醇钠;所述催化剂用量为2
‑
氯
‑4‑
硝基苯
‑
2,3,4
‑
三乙酰基
‑
α
‑
l
‑
岩藻糖苷的0.5
‑
5mol%,优选1
‑
2mol%。
[0024]
在本发明的一种实施方式中,步骤(3)中,所述脱乙酰基反应的反应温度为0
‑
50
℃,优选20
‑
25℃,反应时间为0.5
‑
2h。
[0025]
在本发明的一种实施方式中,步骤(3)中,所述脱乙酰基反应所用的溶剂为甲醇。
[0026]
在本发明的一种实施方式中,步骤(3)中,反应后处理采用重结晶处理,即可得到最终产品,重结晶所用溶剂选自乙醇、异丙醇中的一种或几种。
[0027]
本发明还提供了上述方法在生物检测领域中的应用。
[0028]
本发明相对于现有技术,具有以下的优点和效果:
[0029]
与以往的合成方法相比,本发明选用的起始原料l
‑
岩藻糖价格相对便宜,第二步糖苷化反应采用三氟化硼乙醚催化,相比原有技术,大大降低了2
‑
氯
‑4‑
硝基苯酚使用量以及降低了反应温度,不仅节省了成本,也降低了后处理的难度;本发明反应步骤相对较短,且每步反应收率可观,后处理过程未用到柱层析,可实现工业化生;;终产品2
‑
氯
‑4‑
硝基苯
‑
α
‑
l
‑
岩藻糖苷品质较好;总的工艺路线成本降低;因此,该制备方法具有很好的工业化生产的前景。
附图说明
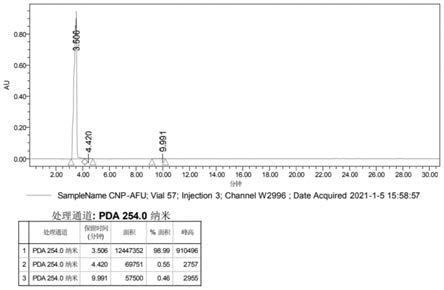
[0030]
图1 2
‑
氯
‑4‑
硝基苯
‑
α
‑
l
‑
岩藻糖苷1h nmr。
[0031]
图2 2
‑
氯
‑4‑
硝基苯
‑
α
‑
l
‑
岩藻糖苷hplc。
具体实施方式
[0032]
下面结合实施例对本发明作进一步的描述,但本发明的实施方式不限于此。
[0033]
实施例1:
[0034]
(1)1,2,3,4
‑
四乙酰基
‑
α
‑
l
‑
岩藻糖的合成
[0035]
往反应器中,加入100g l
‑
岩藻糖和1l吡啶,搅拌;25℃下分批加入乙酸酐746g,防止剧烈放热;加料完毕后,25℃反应12h。反应结束,旋除吡啶得到粘稠状液体;并将粘稠状液体倒入1l二氯甲烷中,水洗(5x 500ml),再用500ml饱和氯化钠溶液洗,用无水硫酸钠干燥后,旋除溶剂,得到1,2,3,4
‑
四乙酰基
‑
α
‑
l
‑
岩藻糖200g,收率为99%。
[0036]
(2)2
‑
氯
‑4‑
硝基苯
‑
2,3,4
‑
三乙酰基
‑
α
‑
l
‑
岩藻糖苷的合成
[0037]
往反应器中,加入200g 1,2,3,4
‑
四乙酰基
‑
α
‑
l
‑
岩藻糖,溶于1l二氯甲烷,搅拌。加入232g2
‑
氯
‑4‑
硝基苯酚和60g三乙胺,氮气保护,搅拌。25℃下,滴加426g bf3·
et2o,滴毕,继续反应48h,停止反应。反应结束,加入1l水淬灭反应,再加入1l二氯甲烷分液。有机相,用饱和氢氧化钠水溶液洗3
×
1l,过滤,分液。合并水相,用1l二氯甲烷反洗一遍。合并有机相,旋除溶剂,得到粗产物2
‑
氯
‑4‑
硝基苯
‑
2,3,4
‑
三乙酰基
‑
α
‑
l
‑
岩藻糖苷,乙醇重结晶,得2
‑
氯
‑4‑
硝基苯
‑
2,3,4
‑
三乙酰基
‑
α
‑
l
‑
岩藻糖苷110g,收率为41%。
[0038]
(3)2
‑
氯
‑4‑
硝基苯
‑
α
‑
l
‑
岩藻糖苷的合成
[0039]
往反应器中,加入100g 2
‑
氯
‑4‑
硝基苯
‑
2,3,4
‑
三乙酰基
‑
α
‑
l
‑
岩藻糖苷,500ml甲醇,室温下搅拌。将125mg甲醇钠溶于5ml甲醇,滴加至反应体系中,搅拌。tlc监测原料反应完全,停止反应。反应结束,加入+h离子交换树脂30g,搅拌淬灭反应。过滤,旋除溶剂,得粗产物5。加入100ml乙醇,打浆,过滤得终产物60g,收率84%。
[0040]
经过测定,终产品2
‑
氯
‑4‑
硝基苯
‑
α
‑
l
‑
岩藻糖苷的品质(纯度)为99%。
[0041]
本实施例通过多次重复实验,实验结果波动不大;而原有技术(日本专利(jp 06
‑
179690)公开的合成方法),多次重复试验发现,实验结果波动较大。
[0042]
实施例2
[0043]
1,2,3,4
‑
四乙酰基
‑
α
‑
l
‑
岩藻糖的合成
[0044]
(1)往反应器中,依次加入3kg乙酸酐和600g无水醋酸钠,加热至80℃,搅拌。分批加入l
‑
岩藻糖1kg,防止剧烈放热;反应放热,控制内温不超过120℃。加料完毕,将内温稳定在100
‑
110℃,反应4h,停止反应。将反应体系降温,加入5l冰水,搅拌过夜。用二氯甲烷3
×
3l将产物萃出,水洗2
×
2l,再用2l饱和氯化钠溶液洗涤一次,有机相用无水硫酸钠干燥处理,旋除溶剂,得到1,2,3,4
‑
四乙酰基
‑
α
‑
l
‑
岩藻糖2kg,收率为99%;
[0045]
(2)2
‑
氯
‑4‑
硝基苯
‑
2,3,4
‑
三乙酰基
‑
α
‑
l
‑
岩藻糖苷的合成
[0046]
往反应器中,加入2kg 1,2,3,4
‑
四乙酰基
‑
α
‑
l
‑
岩藻糖,溶于10l二氯甲烷,搅拌。加入1160g2
‑
氯
‑4‑
硝基苯酚和600g三乙胺,氮气保护,搅拌。25℃下,滴加4.26kg bf3·
et2o,滴毕,继续反应48h,停止反应。反应结束,加入10l水淬灭反应,再加入10l二氯甲烷分液。有机相,用饱和氢氧化钠水溶液洗3
×
10l,过滤,分液。合并水相,用1l二氯甲烷反洗一遍。合并有机相,旋除溶剂,得到粗产物2
‑
氯
‑4‑
硝基苯
‑
2,3,4
‑
三乙酰基
‑
α
‑
l
‑
岩藻糖苷,乙醇重结晶,得2
‑
氯
‑4‑
硝基苯
‑
2,3,4
‑
三乙酰基
‑
α
‑
l
‑
岩藻糖苷1060g,收率为40%。
[0047]
(3)2
‑
氯
‑4‑
硝基苯
‑
α
‑
l
‑
岩藻糖苷的合成
[0048]
往反应器中,加入1000g 2
‑
氯
‑4‑
硝基苯
‑
2,3,4
‑
三乙酰基
‑
α
‑
l
‑
岩藻糖苷,5000ml甲醇,室温下搅拌。将1250mg甲醇钠溶于50ml甲醇,滴加至反应体系中,搅拌。tlc监测原料反应完全,停止反应。反应结束,加入+h离子交换树脂300g,搅拌淬灭反应。过滤,旋除溶剂,得粗产物5。加入1000ml乙醇,打浆,过滤得终产物580g,收率81%。
[0049]
经过测定,终产品2
‑
氯
‑4‑
硝基苯
‑
α
‑
l
‑
岩藻糖苷的品质(纯度)为99%。
[0050]
实施例3
[0051]
(1)1,2,3,4
‑
四乙酰基
‑
α
‑
l
‑
岩藻糖的合成
[0052]
往反应器中,加入100g l
‑
岩藻糖和1l吡啶,搅拌;25℃下分批加入乙酸酐746g,防止剧烈放热;加料完毕后,60℃下反应4h。反应结束,降温,旋除吡啶得到粘稠状液体;并将粘稠状液体倒入1l二氯甲烷中,水洗(5x 500ml),再用500ml饱和氯化钠溶液洗,用无水硫酸钠干燥后,旋除溶剂,得到1,2,3,4
‑
四乙酰基
‑
α
‑
l
‑
岩藻糖;
[0053]
(2)2
‑
氯
‑4‑
硝基苯
‑
2,3,4
‑
三乙酰基
‑
α
‑
l
‑
岩藻糖苷的合成
[0054]
往反应器中,加入200g 1,2,3,4
‑
四乙酰基
‑
α
‑
l
‑
岩藻糖,溶于1l二氯甲烷,搅拌,加入232g2
‑
氯
‑4‑
硝基苯酚和60g三乙胺,氮气保护,搅拌。40℃下,滴加450gtmsotf,滴毕,继续反应48h,停止反应。反应结束,加入1l水淬灭反应,再加入1l二氯甲烷分液。有机相,用饱和氢氧化钠水溶液洗3
×
1l,过滤,分液。合并水相,用1l二氯甲烷反洗一遍。合并有机相,旋除溶剂,得到粗产物2
‑
氯
‑4‑
硝基苯
‑
2,3,4
‑
三乙酰基
‑
α
‑
l
‑
岩藻糖苷,异丙醇重结晶,得2
‑
氯
‑4‑
硝基苯
‑
2,3,4
‑
三乙酰基
‑
α
‑
l
‑
岩藻糖苷;
[0055]
(3)2
‑
氯
‑4‑
硝基苯
‑
α
‑
l
‑
岩藻糖苷的合成
[0056]
往反应器中,加入100g 2
‑
氯
‑4‑
硝基苯
‑
2,3,4
‑
三乙酰基
‑
α
‑
l
‑
岩藻糖苷,500ml甲醇,室温下搅拌。将150mg叔丁醇钠溶于5ml甲醇,滴加至反应体系中,搅拌。tlc监测原料反应完全,停止反应。反应结束,加入+h离子交换树脂30g,搅拌淬灭反应。过滤,旋除溶剂,得粗产物5。加入100ml异丙醇,打浆,过滤得终产物。
[0057]
经过测定,终产品2
‑
氯
‑4‑
硝基苯
‑
α
‑
l
‑
岩藻糖苷的品质(纯度)为99%。
[0058]
对比例1
[0059]
步骤(1)同实施例1;
[0060]
步骤(2):在200ml带有搅拌器三口烧瓶中,加入4.2g无水氯化锌(催化剂),10g 1,2,3,4
‑
四乙酰基
‑
α
‑
l
‑
岩藻糖,52g2
‑
氯
‑4‑
硝基苯酚,加料完毕后,搅拌升温至120℃,待反应液溶清,通过tlc监测反应进程。原料反应完全,停止反应,降温,加入250ml二氯甲烷将反应液溶解。用0.1mol/l氢氧化钠水溶液洗(3x 500ml),再用饱和氯化钠溶液洗(3x 500ml),用无水硫酸钠干燥后,旋除溶剂;通过柱层析(ea/pe=1:5)得到粗产物,再用乙醇重结晶得2
‑
氯
‑4‑
硝基苯
‑
2,3,4
‑
三乙酰基
‑
α
‑
l
‑
岩藻糖苷3g,收率23%。
[0061]
步骤(3)同实施例1;
[0062]
对比例2
[0063]
步骤(1)同实施例1;
[0064]
步骤(2):在2l带有搅拌器三口烧瓶中,加入83g无水氯化锌,200g 1,2,3,4
‑
四乙酰基
‑
α
‑
l
‑
岩藻糖,520g2
‑
氯
‑4‑
硝基苯酚,加料完毕后,搅拌升温至120℃,待反应液溶清,通过tlc监测反应进程。原料反应完全,停止反应,降温,加入5l二氯甲烷将反应液溶解。用0.1mol/l氢氧化钠水溶液洗(3x 1l),再用饱和氯化钠溶液洗(3x2l),用无水硫酸钠干燥后,旋除溶剂;通过柱层析(ea/pe=1:5)得到粗产物,再用乙醇重结晶得2
‑
氯
‑4‑
硝基苯
‑
2,3,4
‑
三乙酰基
‑
α
‑
l
‑
岩藻糖苷48g,收率18%。
[0065]
步骤(3)同实施例1。
[0066]
虽然本发明已以较佳实施例公开如上,但其并非用以限定本发明,任何熟悉此技术的人,在不脱离本发明的精神和范围内,都可做各种的改动与修饰,因此本发明的保护范围应该以权利要求书所界定的为准。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1