一种盐酸马尼地平原料药的合成工艺的制作方法
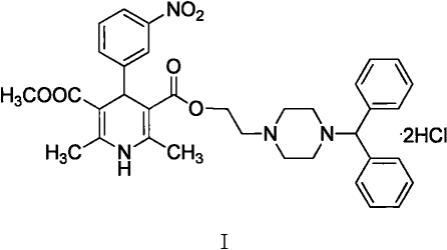
1.本发明属于医药技术领域,具体的涉及一种盐酸马尼地平原料药的合成工艺。
背景技术:
2.盐酸马尼地平,化学名为1,4-二氢-2,6-二甲基-4-(3-硝基苯基)-3,5-吡啶二羧酸2-[4-(二苯甲基)-1-哌嗪基]乙基甲酯二盐酸盐,是一种亲脂性的第三代二氢吡啶类且兼有l型、t型的钙离子双通道阻滞剂,结构式如式i所示。该药具有较强的松弛动脉平滑肌、扩张血管、降低外周血管阻力和动脉压的作用,主要用于治疗轻到中度原发性高血压,对低肾素型高血压的降压效果更明显,并能改善尿酸代谢,对伴有肾衰的高血压患者也可获得很好的效果,对于高血压合并ii型糖尿病患者或者糖耐量降低的患者以及老年患者均具有良好的降压作用,是理想的一线抗高血压用药。
[0003][0004]
专利jp06199789、ep138505、ep94159、wo8304023均提供了一种合成盐酸马尼地平的合成工艺,但都存在不同的弊端,尤其是柱层析法提纯是不适合工业化生产的,批量难以放大,成本居高不下。且中间体和产品纯度也不高,为原料药的质量控制带来较大的风险。
[0005]
中国专利20120097005.1报道,以1-二苯基甲基-4-(2-羟基乙基)哌嗪为起始原料,后续经三步反应得到产品。在盐酸马尼地平制备中先用盐酸成盐,再碱调制备马尼地平游离碱,然后盐酸/甲醇溶液成盐,操作步骤繁琐,采用盐酸/甲醇溶液成盐,使体系内引入了水,导致所得终产物可能为盐酸马尼地平二盐酸盐水合物,难以得到高含量的盐酸马尼地平。
[0006]
本发明通过革新反应路线和物料,摒弃使用双乙烯酮所带来的安全、工艺和健康风险,使反应更温和可控,同时严格控制每步反应参数并对每步反应中间体纯度和杂质都加以控制,尤其是在对潜在基因毒性杂质2-氯乙醇、双乙烯酮、间硝基苯甲醛等的残留都会在工艺中加以控制,其中减压蒸馏并计时保持除去2-氯乙醇残留,使用2,2,6-三甲基-4h-1,3-二英-4-酮替代双乙烯酮,温水洗涤可完全除去间硝基苯甲醛残留,也正是因为在工艺前端就能很好的控制杂质,在成品时就会很容易就得到了高纯度的盐酸马尼地平原料药。使用该工艺得到的产品,纯度高,杂质少,最大单杂为0.05%以下,其他杂质在0.02%以下,无潜在基因毒杂质残留,杂质个数不超过3个,纯度高达99.9%以上。
技术实现要素:
[0007]
本发明的目的是提供一种盐酸马尼地平原料药的合成工艺,本方法操作安全,产品纯度高,尤其中间体和成品杂质控制较好,收率高,适合工业化大生产。
[0008]
为此,本发明采用的技术方案如下:一种盐酸马尼地平原料药原料药的合成工艺,包括以下步骤:
[0009]
①
:将2-氯乙醇加入反应瓶中搅拌,控温开始滴加2,2,6-三甲基-4h-1,3-二英-4-酮,滴加完毕,升温并保温反应,减压浓缩至真空表读数为-0.095mpa并计时保持至蒸尽,得4-氯乙酰乙酸乙酯;
[0010]
②
:将
①
所得的油状物溶解于反应溶剂中并搅拌均匀,加入间硝基苯甲醛和催化剂,开启加热,控温反应,反应结束后,减压浓缩至尽,加入乙酸乙酯和温水搅拌洗涤,分去水相,干燥剂干燥,抽滤,滤液减压浓缩,降温析晶,保温析晶,抽滤,烘干,得2-(3-硝基苯亚甲基)乙酰乙酸氯乙酯固体;
[0011]
③
:将
②
所得的固体溶解于反应溶剂中并搅拌均匀,加入3-氨基巴豆酸甲酯,开启加热,控温反应,反应结束后,减压浓缩,降温析晶,保温析晶,抽滤,烘干,得2,6-二甲基-4-间硝基苯基-1,4-二氢吡啶-3,5-二羧酸甲酯氯乙酯固体;
[0012]
④
:将
③
所得的固体和n-二苯甲基哌嗪加入反应瓶中,加入反应溶剂,开启搅拌和加热,保温反应,反应结束后,降温,控温滴加浓盐酸,保温析晶,再降温析晶,离心。粗品加入醇溶剂中加热溶解,降温析晶,再保温析晶,离心,烘干,即得盐酸马尼地平。
[0013]
所述
①
中,2-氯乙醇与2,2,6-三甲基-4h-1,3-二英-4-酮的质量比为1∶(1.4-2.2),优选为1∶(1.6~2.0);控温温度为0~30℃,优选为10~20℃;保温反应温度为20~80℃,优选为30~60℃;保温反应时间为3~8小时,优选为4~6小时;计时保持时间为1~12小时,优选为3~6小时。
[0014]
所述的
②
中,反应溶剂为甲醇、乙醇、异丙醇、叔丁醇、甲苯、冰醋酸中的一种;催化剂为二甲氨基吡啶、吡啶、哌啶、三乙胺、n,n-二异丙基乙胺中的一种;反应温度为50~110℃,优选为60~100℃;保温反应时间为4~12小时,优选为5~8小时;4-氯乙酰乙酸乙酯∶间硝基苯甲醛∶催化剂∶反应溶剂∶乙酸乙酯∶水的质量比为1∶(0.8~1.2)∶(0.5~1.5)∶(2~6)∶(2~6)∶(2~6),优选为1∶(0.9~1.1)∶(0.8~1.2)∶(3~5)∶(3~5)∶(3~5);温水温度40℃~70℃,优选为50~60℃;为保温析晶温度为-10℃~30℃,优选为-5℃~10℃;析晶时间1~12小时,优选为2~8小时;烘干温度为30℃~90℃,优选为50~65℃;烘干时间为1~10小时,优选为3~6小时。
[0015]
所述的
③
中,2-(3-硝基苯亚甲基)乙酰乙酸氯乙酯∶3-氨基巴豆酸甲酯∶反应溶剂的质量比为1∶(0.28~0.70)∶(2~6),优选为1∶(0.32~0.55)∶(3~5);溶剂为甲醇、乙醇、异丙醇、苯、甲苯、冰醋酸中的一种;反应温度为50~110℃,优选为60~100℃;保温反应时间为4~12小时,优选为5~8小时;保温析晶温度为-10℃~30℃,优选为-5℃~10℃;析晶时间1~12小时,优选为2~8小时;烘干温度为50℃~110℃,优选为75~95℃;烘干时间为3~12小时,优选为4~8小时。
[0016]
所述的
④
中,2,6-二甲基-4-间硝基苯基-1,4-二氢吡啶-3,5-二羧酸甲酯氯乙酯∶n-二苯甲基哌嗪∶反应溶剂的质量比为1∶(0.5~0.8)∶(2~6),优选为1∶(0.6~0.7)∶(3~5);反应溶剂为甲醇、乙醇、异丙醇、叔丁醇、甲苯、冰醋酸中的一种;醇溶剂为甲醇、乙醇、异
丙醇、叔丁醇中的一种;反应温度为50~110℃,优选为60~100℃;保温反应时间为4~12小时,优选为5~8小时;保温析晶温度为-10℃~30℃,优选为-5℃~10℃;析晶时间1~12小时,优选为2~8小时;烘干温度为50℃~100℃,优选为70~85℃;烘干时间为4~18小时,优选为6~12小时。
[0017]
本发明核心是提供一种盐酸马尼地平原料药的合成工艺,本方法操作安全,产品纯度高,尤其中间体和成品杂质控制较好,收率高,适合工业化大生产。本发明通过革新反应路线和物料,摒弃使用双乙烯酮所带来的安全、工艺和健康风险,使反应更温和可控,同时严格控制每步反应参数并对每步反应中间体纯度和杂质都加以控制,尤其是在对潜在基因毒性杂质2-氯乙醇、双乙烯酮、间硝基苯甲醛等的残留都会在工艺中加以控制,其中减压蒸馏并计时保持除去2-氯乙醇残留,使用2,2,6-三甲基-4h-1,3-二英-4~酮替代双乙烯酮,温水洗涤可完全除去间硝基苯甲醛残留。也正是因为在工艺前端就能很好的控制杂质,在成品时就会很容易就得到了高纯度的盐酸马尼地平原料药。使用该工艺得到的产品,纯度高,杂质少,最大单杂为0.05%以下,其他杂质在0.02%以下,无潜在基因毒杂质残留,杂质个数不超过3个,纯度高达99.9%以上。
具体实施方式
[0018]
为了更好地理解本发明,下面将通过本发明的实施例对本发明进行详细的描述和说明,本发明并不局限于下面的实施例,这些实施例不以任何方式限制本发明的范围。本领域的技术人员在权利要求的范围内所做出的某些改变和调整也应认为属于本发明的范围。
[0019]
实施例1
[0020]
将161.4g(2.0mol)2-氯乙醇加入反应瓶中搅拌,控温15℃开始滴加284.3g(2.0mol)2,2,6-三甲基-4h-1,3-二英-4-酮,滴加完毕,升温至60℃并保温反应5小时,减压浓缩至真空表读数为-0.095mpa并计时保持3小时至蒸尽,得325.7g 4-氯乙酰乙酸乙酯,纯度99.1%,2-氯乙醇残留0.008%,收率99.1%;
[0021]
将325.7g(1.98mol)4-氯乙酰乙酸乙酯溶解于1303g甲苯中并搅拌均匀,加入287.1g(1.9mol)间硝基苯甲醛和402.8g(3.1mol)n,n-二异丙基乙胺,开启加热,控温110℃反应8小时,反应结束后,减压浓缩至尽,加入1303g乙酸乙酯和1303g 50℃温水搅拌洗涤,分去水相,干燥剂干燥,抽滤,滤液减压浓缩至1000g,降温析晶,10℃保温析晶8小时,抽滤,60℃烘干6小时,得550g 2-(3-硝基苯亚甲基)乙酰乙酸氯乙酯固体,纯度96.9%,间硝基苯甲醛残留0.01%,收率90.5%;
[0022]
将550g(1.79mol)2-(3-硝基苯亚甲基)乙酰乙酸氯乙酯固体溶解于1650g甲醇中并搅拌均匀,加入208.4g(1.81mol)3-氨基巴豆酸甲酯,开启加热,控温80℃反应6小时,反应结束后,减压浓缩至1600g,降温至5℃析晶,保温5℃析晶12小时,抽滤,90℃烘干6小时,得650.9g 2,6-二甲基-4-间硝基苯基-1,4-二氢吡啶-3,5-二羧酸甲酯氯乙酯固体,纯度98.5%,收率90.8%;
[0023]
将650.9g(1.62mol)2,6-二甲基-4-间硝基苯基-1,4-二氢吡啶-3,5-二羧酸甲酯氯乙酯固体和449.7g(1.78mol)n-二苯甲基哌嗪加入反应瓶中,加入2604g叔丁醇,开启搅拌和加热,80℃保温反应12小时,反应结束后,降温,控温20℃滴加浓盐酸至ph=2,保温20℃析晶,再降温至0℃析晶12小时,离心。粗品加入3900g异丙醇中加热溶解,降温至30℃析
晶6小时,再降温至0℃析晶12小时,离心,70℃烘干12小时,即得盐酸马尼地平888.7g。收率:80.2%,熔点206.1℃~210.7℃,最大单杂0.05%,总杂为0.08%。总收率65.0%。
[0024]
实施例2
[0025]
将161.4g(2.0mol)2-氯乙醇加入反应瓶中搅拌,控温30℃开始滴加298.5g(2.1mol)2,2,6-三甲基-4h-1,3-二英-4-酮,滴加完毕,升温至80℃并保温反应6小时,减压浓缩至真空表读数为-0.095mpa并计时保持6小时至蒸尽,得323.6g 4-氯乙酰乙酸乙酯,纯度99.0%,2-氯乙醇残留0.005%,收率97.4%;
[0026]
将323.6g(1.95mol)4-氯乙酰乙酸乙酯溶解于1295g冰醋酸中并搅拌均匀,加入287.1g(1.9mol)间硝基苯甲醛和264g(3.1mol)哌啶,开启加热,控温100℃反应12小时,反应结束后,减压浓缩至尽,加入1295g乙酸乙酯和1295g 60℃温水搅拌洗涤,分去水相,干燥剂干燥,抽滤,滤液减压浓缩至900g,降温析晶,0℃保温析晶12小时,抽滤,70℃烘干8小时,得545.4g 2-(3-硝基苯亚甲基)乙酰乙酸氯乙酯固体,纯度97.3%,间硝基苯甲醛残留0.009%,收率91.5%;
[0027]
将545.4g(1.78mol)2-(3-硝基苯亚甲基)乙酰乙酸氯乙酯固体溶解于1636g异丙醇中并搅拌均匀,加入207.2g(1.80mol)3-氨基巴豆酸甲酯,开启加热,控温82℃反应12小时,反应结束后,减压浓缩至1500g,降温至0℃析晶,保温0℃析晶12小时,抽滤,75℃烘干12小时,得641.1g 2,6-二甲基-4-间硝基苯基-1,4-二氢吡啶-3,5-二羧酸甲酯氯乙酯固体,纯度98.8%,收率90.2%;
[0028]
将641.1g(1.60mol)2,6-二甲基-4-间硝基苯基-1,4-二氢吡啶-3,5-二羧酸甲酯氯乙酯固体和484.5g(1.92mol)n-二苯甲基哌嗪加入反应瓶中,加入1923g异丙醇,开启搅拌和加热,75℃保温反应6小时,反应结束后,降温,控温30℃滴加浓盐酸至ph=2,保温30℃析晶,再降温至-5℃析晶8小时,离心。粗品加入2500g乙醇中加热溶解,降温至20℃析晶12小时,再降温至-5℃析晶12小时,离心,55℃烘干12小时,即得盐酸马尼地平921.4g。收率:84.2%,熔点206.1℃~210.7℃,最大单杂0.05%,总杂为0.08%,总收率67.4%。
[0029]
本发明的有益效果如下:本发明通过革新反应路线和物料,摒弃使用双乙烯酮所带来的安全、工艺和健康风险,使反应更温和可控,同时严格控制每步反应参数并对每步反应中间体纯度和杂质都加以控制,尤其是在对潜在基因毒性杂质2-氯乙醇、双乙烯酮、间硝基苯甲醛等的残留都会在工艺中加以控制,其中减压蒸馏并计时保持除去2-氯乙醇残留,使用2,2,6-三甲基-4h-1,3-二英-4-酮替代双乙烯酮,温水洗涤可完全除去间硝基苯甲醛残留。也正是因为在工艺前端就能很好的控制杂质,在成品时就会很容易就得到了高纯度的盐酸马尼地平原料药。使用该工艺得到的产品,纯度高,杂质少,最大单杂为0.05%以下,其他杂质在0.02%以下,无潜在基因毒杂质残留,杂质个数不超过3个,纯度高达99.9%以上。
[0030]
最后应说明的是,以上实施例仅用以说明本发明的技术方案,而非对其限制,尽管参照上述实施例对本发明进行了详细的说明,所属领域的普通技术人员应当理解,依然可以对本发明的具体实施方式进行修改或者等同替换,而未脱离本发明精神和范围的任何修改或者等同替换,其均涵盖在本发明的权利要求范围当中。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1