一种二氮杂桥化合物的合成方法与流程
1.本发明涉及有机合成领域,具体涉及一种二氮杂桥化合物的合成方法。
背景技术:
2.二氮杂桥化合物是一种非常有用的医药中间体,有大量的药物具有二氮杂 桥化合物的片段,如美国礼来公司开发的一种用于治疗转染重排基因融合阳性 的转移性非小细胞肺癌的药物selpercatinib(化合物i),美国爱斯凯利尔公司 开发的一种视黄酸相关的孤儿核受体调节剂(化合物ii),美国先灵公司开发的 一种用于抑制i型11β-羟基类固醇脱氢酶的药物(化合物iii)以及德国拜耳公 司开发的用于治疗子宫内膜异位的药物(化合物iv)等等。
[0003][0003][0004]
根据文献报道,上述药物在合成的过程中均直接或间接地涉及到化合物1a 或2a。
[0005][0006]
在现有技术中,化合物1a的合成方法如下:
[0007][0008]
由上述反应路线可知,从化合物v到化合物1需要经过两步,并且还会额 外产生大量的邻硝基苯磺酸,邻硝基甲苯磺酸的释出不仅会给反应的后处理带 来麻烦,而且会使整个反应体系呈酸性,这可能会使得叔丁基氧羰基脱落从而 降低反应收率。
[0009]
进一步地,上述路线是以化合物cis-2为起始原料,然而化合物cis-2在合 成过程中会产生大量的反式副产物,且顺反比约为1:1,显然这样会导致收率 偏低。由于产物顺反比较低,所以后续需要通过柱层析的方式对顺反两种产物 进行分析,这也使得化合物1a的生产难以工艺化。
[0010]
在现有技术中,化合物2a的合成方法如下:
[0011][0012]
由上述路线可知,在化合物2a的合成过程中不仅需要经历230℃高温这样 较为苛刻的反应条件,还需要反复多次使用pd/c这样的还原试剂,整个路线 较长,实际生产过程中不够经济环保。
[0013]
此外,该路线还对起始反应底物化合物vii的立体构型有一定的要求,这 会进一步增加生产的成本。
技术实现要素:
[0014]
本发明是为了解决上述问题而进行的,目的在于提供一种步骤简化、收率 高的二氮杂桥化合物的合成方法。
[0015]
除非另外指明,否则如本文所使用,以下定义应适用。另外,本文所定义 的许多基团可以任选被取代。定义中的取代基列举是示例性的并且不应解释为 限制在本说明书别处所定义的取代基。
[0016]
在本文中,术语“烷基”是指不含不饱和度并且具有一至十个碳原子的仅 由碳原子和氢原子组成的直链或分支链烃链基团,该基团经由单键附接到分子 其余部分,例如甲基、乙基、正丙基、1-甲基乙基(异丙基)、正丁基、正戊基 及1,1-二甲基乙基(叔丁基)。术语“c1-10烷基”是指具有最多10个碳原子的 如以上所定义的烷基。
[0017]
在本文中,术语“芳基”是指碳原子数在6到20个范围内的芳香族基团, 如苯基、萘基、四氢萘基、茚满基及联苯基等。
[0018]
在本文中,术语“取代”是指被以下取代基中的任一个或任何组合取代, 这些取代基可以相同或不同并且独立地选自烷基、羟基、卤素、羧基、氰基、 硝基,如术语“取代芳基”可以指代对甲基苯基、邻硝基苯基等。
[0019]
在本文中,术语“卤代”、“卤化物”或替代地,“卤素”意思指氟、氯、 溴或碘。术语“卤代烷基”指代包括被一个或多个卤代基或其组合取代的烷基, 如三氟甲基等。
[0020]
在本文中所使用的术语“接触”应做广义理解,其可以是任何能够使得至 少两种反应物发生化学反应的方式,例如可以是将两种反应物在适当的条件下 进行混合。根据需要,可以在搅拌下,将需要进行接触的反应物进行混合,由 此,搅拌的类型并不受特别限制,例如可以为机械搅拌,即在机械力的作用下 进行搅拌。
[0021]
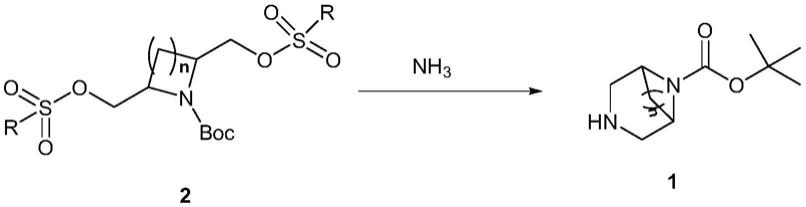
本发明提供了一种二氮杂桥化合物的合成方法,具有这样的特征,反应方 程式为:
[0022][0023]
式中,r为芳基、取代芳基、烷基或卤代烷基,n=1或2,
[0024]
反应步骤如下:步骤1,将化合物2与nh3或nh3溶液接触,得到反应混 合物;步骤2,对反应混合物进行后处理,即得化合物1。
[0025]
在本发明提供的二氮杂桥化合物的合成方法中,还可以具有这样的特征: 其中,在步骤1中,将化合物2与nh3或nh3溶液在反应媒介中接触。
[0026]
在本发明提供的二氮杂桥化合物的合成方法中,还可以具有这样的特征: 其中,在步骤1中,使化合物2与nh3或nh3溶液接触的方法,可以是化合物 2或其溶液加入到nh3或其溶液中,也可以是nh3或nh3溶液加入到化合物2 或其溶液中,nh3或nh3溶液可以是氨气、液氨、氨水或者氨的有机溶液。
[0027]
在本发明提供的二氮杂桥化合物的合成方法中,还可以具有这样的特征: 其中,反应媒介为非醇类液体。
[0028]
在本发明提供的二氮杂桥化合物的合成方法中,还可以具有这样的特征: 其中,非醇类液体为水、乙腈、dmf或四氢呋喃中的任意一种或多种。
[0029]
在本发明提供的二氮杂桥化合物的合成方法中,还可以具有这样的特征: 其中,化合物2中的r为甲基或对甲基苯基。
[0030]
在本发明提供的二氮杂桥化合物的合成方法中,还可以具有这样的特征: 其中,在步骤1中,化合物2与nh3或nh3溶液在25℃-80℃接触,优选为 30℃-70℃。
[0031]
在本发明提供的二氮杂桥化合物的合成方法中,还可以具有这样的特征: 其中,在步骤1中,nh3溶液为浓度为15wt%-35wt%的氨水,优选为浓度为 25wt%-28wt%的氨水。
[0032]
在本发明提供的二氮杂桥化合物的合成方法中,还可以具有这样的特征: 其中,步骤2中后处理的包括如下步骤:向反应混合物中加入萃取剂,萃取, 取有机相,水洗,重结晶,即得化合物1,其中,重结晶步骤中使用的重结晶 剂为为乙酸乙酯与石油醚体积比为1:(8-20)的混合液,优选为乙酸乙酯与石 油醚体积比为1:(8-11)的混合液。
[0033]
在本发明提供的二氮杂桥化合物的合成方法中,还可以具有这样的特征: 其中,化合物2的结构式为
[0034][0035]
在本发明提供的二氮杂桥化合物的合成方法中,还可以具有这样的特征: 其中,化合物2制备的反应方程式为:
[0036][0037]
式中,r’为c1-c10的烷基,
[0038]
包括如下步骤:步骤a,将化合物3与苄胺接触,得到化合物4;步骤b, 将化合物4与酯还原剂接触,得到化合物5;步骤c,将化合物5依次或同时 与苄基脱除剂以及(boc)2o接触,得到化合物6;步骤d,将化合物6与磺酰 化试剂接触,得到化合物2,其中,酯还原剂为可以将酯还原成醇的试剂,该 试剂可以由一种化合物组成也可以由多种化合物组成的。在本发明中,酯还原 剂为用于将酯基还原成羟基的试剂,可以是单一的金属复氢化合物(如氢化铝 锂)、金属复氢化合物与lewis酸组成的复合还原剂(如硼氢化钠/三氯化铝), 也可以是钠单质,苄基脱除剂为将苄基从化合物5从脱除而不影响其他官能团 的试剂,该试剂可以由一种化合物组成如三氟乙酸、ddq(2,3-二氯-5,6-二氰基
ꢀ‑
1,4对苯醌)、硝酸铈铵,也可以由多种化合物组成,如h2/pd/c、na/液氨/叔 丁醇等,磺酰化试剂为可以与化合物6反应形成磺酸酯的试剂,可以是磺酰氯 (rso2cl)、磺酰溴(rso2br)、磺酸酐等。
[0039]
在本发明提供的二氮杂桥化合物的合成方法中,还可以具有这样的特征: 其中,在步骤a中,在缚酸剂的存在下,化合物3与苄胺接触,缚酸剂为二异 丙基乙基胺或三乙胺中的任意一种或两者的混合物。
[0040]
在本发明提供的二氮杂桥化合物的合成方法中,还可以具有这样的特征: 其中,在步骤a中,溶剂为dmf、乙腈、甲苯或四氢呋喃中的一种或多种。
[0041]
在本发明提供的二氮杂桥化合物的合成方法中,还可以具有这样的特征: 其中,
在步骤a中,在将化合物4与酯还原剂接触后,还包括后处理操作,后 处理操作包括:向化合物4与酯还原剂接触后得到的反应液中加入萃取剂,萃 取,取有机相,向有机相中通入酸性气体,控温在-10℃~10℃,搅拌1h-5h, 过滤,取固体,干燥,即得化合物5。
[0042]
在本发明提供的二氮杂桥化合物的合成方法中,还可以具有这样的特征: 其中,萃取剂为甲苯与水的混合溶液,优选为甲苯与水体积比为(1-3):(1
‑ꢀ
3)的混合液,萃取剂与反应原液的体积比为1:(0.8-2),酸性气体为盐酸气。
[0043]
在本发明提供的二氮杂桥化合物的合成方法中,还可以具有这样的特征: 其中,化合物2的结构式为
[0044][0045]
制备化合物2反应方程式为:
[0046][0047]
式中,r’为c1-c10的烷基,
[0048]
包括如下步骤:步骤α,将化合物7与芳基还原剂接触,得到化合物8; 步骤β,将化合物8与酯还原剂接触,得到化合物9;步骤γ,将化合物9余 磺酰化试剂接触,得到化合物2,其中,芳基还原剂为用于将吡咯催化加氢还 原成吡咯烷的试剂,如pt/c/h2,cu/al2o3,钌催化剂,镍催化剂等;酯还原剂 为用于将酯基还原成羟基的试剂,可以是单一的金属复氢化合物(如氢化铝锂) 、金属复氢化合物与lewis酸组成的复合还原剂(如硼氢化钠/三氯化铝),也 可以是钠单质;磺酰化试剂为可以与化合物6反应形成磺酸酯的试剂,可以是 磺酰氯(rso2cl)、磺酰溴(rso2br)、磺酸酐等。
[0049]
发明的作用与效果
[0050]
根据本发明所涉及的二氮杂桥化合物的合成方法,因为以化合物2和nh3或nh3的溶液为原料,所以,本发明不仅能够有效缩短工艺流程,节约工艺成 本,还能够在一定程度上提高反应收率。
附图说明
[0051]
图1是本发明的实施例1中6-(叔丁氧羰基)-3,6-二氮杂双环[3.1.1]庚烷的气 相谱图;
[0052]
图2是本发明的实施例1中6-(叔丁氧羰基)-3,6-二氮杂双环[3.1.1]庚烷的核 磁谱图;
[0053]
图3是本发明的实施例3中的顺式-1-苄基-2,4-二烷氧基羰基氮杂环丁烷核 磁谱图;以及
[0054]
图4是本发明的实施例6中3,8-二氮杂双环[3.2.1]辛烷-8-甲酸叔丁酯的核 磁谱图。
具体实施方式
[0055]
为了使本发明实现的技术手段、创作特征、达成目的与功效易于明白了解, 以下结合实施例及附图对本发明作具体阐述。
[0056]
在下述实施例中,hplc检测的条件为:c18柱,紫外波长210nm,柱温 30℃,流速0.8ml/min,进样量10μl,流动相为85vol%的0.3wt%三氟乙酸水 溶液及15vol%乙腈。
[0057]
在下述实施例中,gc检测的条件为:db-624毛细管柱 (30mm
×
0.32mm
×
1.8μm),分流比20:1,载气为氮气,气化室温度为280℃, 检测器温度为280℃,升温程序为初始温度60℃,保持2min,以15℃/min升 至240℃。
[0058]
除另有注明外,下述各实施例中的原料来源均为市售,化学纯。
[0059]
《实施例1》
[0060]
6-(叔丁氧羰基)-3,6-二氮杂双环[3.1.1]庚烷的合成
[0061]
本实施例提供了一种6-(叔丁氧羰基)-3,6-二氮杂双环[3.1.1]庚烷(即化合物1a)的合成方法,反应方程式为:
[0062][0063]
反应步骤如下:
[0064]
步骤1,将化合物cis-2a 5g(13.4mmol,通过实施例5的方法制备而成) 加入到15ml乙腈中,再加入25wt%-28wt%的浓氨水(市售,使用前未经滴定) 50ml,加热至70℃,搅拌反应12小时,得到反应液;
[0065]
步骤2,向反应液中加入20ml二氯甲烷,搅拌,萃取,取有机相,水洗 有机相,加入100ml乙酸乙酯/石油醚混合液进行重结晶,乙酸乙酯/石油醚混 合液为乙酸乙酯与石油醚体积比为1:9的混合液,得1.9g呈类白色固体的化合 物1a(即6-(叔丁氧羰基)-3,6-二氮杂双环[3.1.1]庚烷),收率71.7%。
[0066]
图1是本发明的实施例1中6-(叔丁氧羰基)-3,6-二氮杂双环[3.1.1]庚烷的气 相谱图。
[0067]
如图1所示,化合物1的gc纯度94.8%,无需进一步提纯即可投入后续反 应中。
[0068]
图2是本发明的实施例1中6-(叔丁氧羰基)-3,6-二氮杂双环[3.1.1]庚烷的核 磁
谱图。
[0069]
如图2所示,本实施例得到的6-(叔丁氧羰基)-3,6-二氮杂双环[3.1.1]庚烷 (即化合物1a)的氢谱与文献报道一致。
[0070]
《实施例2》
[0071]
反应条件的筛选
[0072]
本实施例在实施例1的基础上,对反应条件进行了进一步筛选,除表中列 出的条件外,其余反应条件以及操作均与实施例1相同,具体筛选的反应条件 以及相应的反应结果如表1所示。
[0073]
表1化合物1a的合成反应条件筛选表
[0074]
编号溶剂反应温度收率纯度1dmf70℃65.5%83.4%2乙腈30℃51.2%92.8%3dmf30℃45.1%81.4%4thf70℃57.2%78.3%5thf30℃43.3%72.4%6甲醇60℃痕量-[0075]
如表1所示,当使用dmf、乙腈以及四氢呋喃作为反应溶剂时,反应都能 顺利地进行,然而当使用甲醇作为溶剂时,无法得到目标产物,这可能是因为 化合物cis-2a与甲醇发生酯交换反应,而发生酯交换反应后的产物无法与氨水 进行反应,因此也无法得到目标产物。
[0076]
此外,当反应温度为70℃时,相对于反应温度为30℃时,化合物1的收 率明显提高。产物纯度则与溶剂有一定的关系,当溶剂选用乙腈时,产物的纯 度明显优于溶剂选用dmf以及thf时的情形。
[0077]
《实施例3》
[0078]
顺式-1-苄基-2,4-二乙氧基羰基氮杂环丁烷的合成
[0079]
本实施例提供了一种顺式-1-苄基-2,4-二乙氧基羰基氮杂环丁烷的合成方法, 反应方程式为:
[0080][0081]
具体包括如下步骤:
[0082]
步骤1,在250ml反应瓶中加入2,4-二溴戊二酸二乙酯 10.0g(28.9mmol,1eq),苄胺3.1g(28.9mmol,1eq),二异丙基乙基胺 7.5g(58.0mmol,2eq)以及乙腈100ml,加热至85℃回流反应5小时,得反应 原液。此时,对反应原液进行取样,使用hplc进行反应原液进行检测,在反 应原液中顺式产物与反式产物的比例为60.6%:12.3%,即4.9:1。
[0083]
步骤2,将反应原液浓缩至20ml,加入由50ml甲苯和50ml水组成的萃 取剂后进行萃取,取有机相。向有机相中通入盐酸气至ph至为3并控温在0℃, 在通入盐酸气的同时保持搅拌,在搅拌的过程中有机相中逐渐有晶体析出,在 ph调节完成后继续控温在0℃搅拌2
小时,过滤,取固体,烘干,得目标产物 5.82g,为白色固体,收率为69.2%,液相纯度92.8%,可以不经过进一步纯化 直接投入下一步反应中。
[0084]
图3是本发明的实施例中的顺式-1-苄基-2,4-二烷氧基羰基氮杂环丁烷核磁 谱图。
[0085]
如图3所示,与顺式标准样品一致,因此可以确定产物为顺式-1-苄基-2,4
‑ꢀ
二乙氧基羰基氮杂环丁烷。
[0086]
《对照例》
[0087]
顺式-1-苄基-2,4-二乙氧基羰基氮杂环丁烷的合成
[0088]
本对照例提供了一种顺式-1-苄基-2,4-二乙氧基羰基氮杂环丁烷的合成方法, 反应方程式为:
[0089][0090]
具体包括如下步骤:
[0091]
步骤1,在250ml反应瓶中加入2,4-二溴戊二酸二乙酯 10.0g(28.9mmol,1eq),苄胺9.3g(86.7mmol,3eq)以及dmf 100ml,加热至 85℃反应5小时,得反应原液。此时,对反应原液进行取样,使用hplc进行 反应原液进行检测,在反应原液中顺式产物与反式产物的比例为 31.6%:37.6%,即1:0.84。
[0092]
步骤2,将反应原液浓缩至20ml,加入由100ml二氯甲烷和100ml水组 成的萃取剂后进行萃取,取有机相。由于顺反比例较低无法进行重结晶,因此 将有机相浓缩后,进行柱层析分离,得目标产物1.86g,为黄色油状液体,收 率为22.1%。
[0093]
《实施例4》
[0094]
反应条件的筛选
[0095]
本实施例在实施例3的基础上,对反应条件进行了进一步筛选,具体的筛 选条件以及相应的实验结果如表2所示。
[0096]
表2化合物cis-4a的合成反应条件筛选表
[0097]
编号缚酸剂溶剂顺式:反式顺式产品收率1二异丙基乙基胺dmf3.96:156.3%2二异丙基乙基胺甲苯2.66:135.6%3二异丙基乙基胺四氢呋喃
*
2.92:137.5%4三乙胺乙腈3.11:148.3%
[0098]
*
反应温度为70℃回流
[0099]
如表2所示,当缚酸剂选用二异丙基乙基胺或三乙胺时,产品的顺反比例 均高于采用苄胺作为缚酸剂(参见对照例)时的顺反比例;溶剂对于顺反比例 的影响并不是很大,顺式产品的收率均在30%-50%之间,其中,dmf和乙腈的 收率可以达到40%以上。
[0100]
《实施例5》
[0101]
化合物cis-2a的合成
[0102]
本实施例提供了化合物cis-2的制备方法,反应方程式如下:
[0103]
具体步骤 如下:
[0104]
步骤1,在0℃下,将10g化合物cis-4a(34.3mmol,1eq,通过实施例3 的方法制备)溶解在200ml无水乙醚中,加入2.74g lialh4(72.0mmol,2.1eq),0℃下反应4h后滴加20ml水猝灭反应,取有机 相,饱和食盐水洗涤(100ml
×
2次),无水硫酸钠干燥,减压浓缩,得5.69g 化合物cis-5,收率80%;
[0105]
步骤2,将5g化合物cis-5(24.1mmol,1eq)溶解在120ml乙醇中,加入 0.9g pd/c(10%),置换气,充入氢气,室温下搅拌反应36h,加入7.9g(boc) 2
o(36.2mmol,1.5eq)以及6.1g碳酸氢钠(72.3mmo,3eq),室温下继续反应 12h,硅藻土过滤,取滤液,无水硫酸钠干燥,加压浓缩,得4.7g化合物cis
‑ꢀ
6,收率90%;
[0106]
步骤3,将4g化合物cis-6(18.4mmol,1eq)溶解在100ml二氯甲烷中, 在冰水浴下依次滴入3.7g三乙胺(36.8mmol,2eq)以及5.1g mscl(44.2mmol,2.4eq),自然回至室温,反应3h,加入100ml水洗涤,减 压浓缩,加入150ml石油醚打浆,过滤,取固体,60℃干燥6h,即得5.9g化 合物cis-2a,收率86%。
[0107]
《实施例6》
[0108]
化合物3,8-二氮杂双环[3.2.1]辛烷-8-甲酸叔丁酯的合成
[0109]
本实施例提供了一种3,8-二氮杂双环[3.2.1]辛烷-8-甲酸叔丁酯的合成方法, 反应方程式为:
[0110][0111]
具体步骤如下:
[0112]
步骤1,将化合物2b 5g(12.9mmol,通过实施例9的方法制备而成)加入 到15ml乙腈中,再加入25wt%-28wt%的浓氨水(市售,使用前未经滴定) 50ml,加热至70℃,搅拌反应12小时,得到反应液;
[0113]
步骤2,向反应液中加入20ml二氯甲烷,搅拌,萃取,取有机相,水洗 有机相,加入100ml乙酸乙酯/石油醚混合液进行重结晶,乙酸乙酯/石油醚混 合液为乙酸乙酯与石油醚体积比为1:9的混合液,得2.0g呈类白色固体的化 合物2b(即3,8-二氮杂双环[3.2.1]辛烷-8-甲酸叔丁酯),收率72.9%,gc纯度 95.3%,无需进一步提纯即可投入后续反应中。
[0114]
图4是本发明的实施例6中6-(叔丁氧羰基)-3,6-二氮杂双环[3.1.1]庚烷的核 磁谱图。
[0115]
如图4所示,本实施例得到的3,8-二氮杂双环[3.2.1]辛烷-8-甲酸叔丁酯 (即化合物1b)的氢谱与文献报道一致。
[0116]
《实施例7》
[0117]
化合物3,8-二氮杂双环[3.2.1]辛烷-8-甲酸叔丁酯的合成
[0118]
本实施例提供了一种3,8-二氮杂双环[3.2.1]辛烷-8-甲酸叔丁酯的合成方法, 反应方程式为:
[0119][0120]
具体步骤如下:
[0121]
步骤1,将化合物2b 5g(12.9mmol,通过实施例9的方法制备而成)加 入到65ml 0.5m的氨的四氢呋喃溶液中,加热至70℃回流,搅拌反应12小时, 得到反应液;
[0122]
步骤2,向反应液中加入20ml二氯甲烷,搅拌,萃取,取有机相,水洗 有机相,加入100ml乙酸乙酯/石油醚混合液进行重结晶,乙酸乙酯/石油醚混 合液为乙酸乙酯与石油醚体积比为1:9的混合液,得1.60g呈类白色固体的化 合物2b(即3,8-二氮杂双环[3.2.1]辛烷-8-甲酸叔丁酯),收率58.4%,gc纯度 92.5%,无需进一步提纯即可投入后续反应中。
[0123]
《实施例8》
[0124]
本实施例在实施例6的基础上,对反应条件进行了进一步筛选,除表中列 出的条件外,其余反应条件以及操作均与实施例6相同,具体筛选的反应条件 以及相应的反应结果如表3所示。
[0125]
表3化合物1b合成的反应条件筛选表
[0126][0127][0128]
由表3可知,与化合物1a的合成相似,以dmf、乙腈或thf作溶剂,反 应均能顺利进行,但是以甲醇作为溶剂时,反应无法顺利进行。此外,适当地 提高反应温度有利于提高反应的收率以及纯度。
[0129]
《实施例9》
[0130]
化合物2b的合成
[0131]
本实施例提供了一种化合物2b的合成方法,反应方程式为:
[0132]
具体步骤如下:
[0133]
步骤1,将20g化合物7b(70.6mmol,在本实施例中,化合物7b参照论文donohoetj,headleyce,cousinsr,etal.flexibilityinthepartialreductionof2,5-disubstitutedpyrroles:applicationtothesynthesisofdmdp.[j].organicletters,2003,5(7):999-1002.报道的方法合成)溶解在500ml甲醇中,室温下置换氮气,加入6gpt/c(pt含量10%),充入氢气至反应体系内氢气的分压为1atm,室温下搅拌反应8h,使用硅藻土过滤,取滤液,蒸除溶剂,即得19.4g化合物8b,收率95.6%,未经进一步纯化,直接投入下一步;
[0134]
步骤2,在0℃下,将4.8glialh4(126.3mmol,2.0eq)溶解在200ml无水四氢呋喃溶液中,得到lialh4的四氢呋喃溶液,将18g化合物8b(62.6mmol,1.0eq)溶解在300ml无水四氢呋喃中,得到化合物8b的四氢呋喃溶液。在氮气保护下,将化合物8b的四氢呋喃溶液加入到lialh4的四氢呋喃溶液中,室温下搅拌反应20h,加入300ml饱和氯化铵水溶液以及150ml乙酸乙酯猝灭反应,萃取,取有机相,分别用1mhcl水溶液(300ml
×
3)以及饱和食盐水(300ml
×
1)洗涤,无水硫酸钠干燥,减压蒸除溶剂,即得12.4g化合物9,收率85.6%,未经进一步纯化,直接投入下一步;
[0135]
步骤3,将12g化合物9(51.9mmol,1eq)溶解在200ml无水四氢呋喃中,加入13.1g三乙胺(129.8mmol,2.5eq),在冰水浴下,滴加13.1g甲基磺酰氯(114.2mmol,2.2eq),自然回至室温,搅拌反应12h,依次用蒸馏水(300ml
×
1)、10wt%柠檬酸水溶液(300ml
×
1)以及饱和食盐水(300ml
×
1)洗涤,取有机相,无水硫酸钠干燥,减压浓缩,氯仿/正庚烷(v/v=1:10)体系重结晶,得到16.5g化合物2b,收率82.1%,液相纯度98.3%,可以不经进一步提纯直接投入后续反应中。
[0136]
实施例的作用与效果
[0137]
根据上述实施例所涉及的二氮杂桥化合物的合成方法,因为以化合物2a或化合物2b和nh3为原料,所以,本发明不仅能够有效缩短工艺流程,节约工艺成本,还能够在一定程度上提高反应收率。
[0138]
进一步地,因为采用了乙腈作为化合物2a或化合物2b和氨水反应的反应溶剂,所以,可以在保证收率的同时有效提升产物纯度,从而可以使得产物无需进一步提纯直接进行下一步反应。
[0139]
进一步地,因为在制备顺式-1-苄基-2,4-二烷氧基羰基氮杂环丁烷(即cis-4a)
的反应中采用了二异丙基乙基胺或三乙胺作为缚酸剂,以dmf或乙腈为 溶剂,所以,本发明能够以高收率得到较高的顺反比例值的顺式-1-苄基-2,4-二 烷氧基羰基氮杂环丁烷。
[0140]
进一步地,上述实施例还在得到高顺反比例的产物的基础上,开发了一种 适用于二氮杂桥化合物的结晶方法,一方面可以避免柱层析,简化后处理操作, 使得产品工艺化成为可能,另一方面,通过结晶的方式使得最终产品的状态为 固态,方便存储和运输。
[0141]
上述实施方式为本发明的优选案例,并不用来限制本发明的保护范围。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1