一种基于氨基酸-羟基酸共聚物的纳米载药体系及其制备方法和应用
一种基于氨基酸
‑
羟基酸共聚物的纳米载药体系及其制备方法和应用
技术领域
1.本发明属于生物医用材料技术领域,更具体地,涉及一种基于氨基酸和羟基酸共聚物的纳米载药体系及其制备方法和应用。
背景技术:
2.近年来,恶性肿瘤成为威胁人们身心健康的重大疾病,并呈明显上升趋势,对人类健康造成极大的影响。化疗是肿瘤学中最常用的治疗方法之一,蛋白酶类抑制剂近年来在抗肿瘤方面也得到广泛的应用,蛋白酶类抑制剂与化学治疗剂的组合在治疗具有较强转移性的恶性肿瘤方面具有较大的潜力。然而,治疗效果的好坏在很大程度上取决于是否有足够的浓度化疗药物或蛋白酶类抑制剂到达整个肿瘤部位。常规的抗癌药物制剂(如紫杉醇ptx)和蛋白酶类抑制剂(如pf
‑
543)由于其水溶性差和选择性差,常导致不令人满意的治疗效果和严重的毒副作用,如在全身循环中被快速清除并且对癌细胞和健康细胞具有相似的细胞毒性。因此,寻求一种方法来增加抗癌药物制剂的体内循环时间、在保证对肿瘤细胞杀伤作用的同时降低药物对正常细胞的毒性成为关键瓶颈。
3.在医用高分子迅速发展的当下,在提高医用高分子生物相容性的基础上实现其多功能性是生物材料追求的目标。因此,需要开发一种生物相容好、多功能性和可以生物降解的聚合物载体库,来降低疾病风险,改善人类健康。其中,纳米递送系统依赖于实体瘤的高通透性和滞留效应(epr)来改善药代动力学特征和靶位点积累,有望彻底改变肿瘤的诊断和治疗。近年来,文献中报道了许多基于不同的天然材料的药物载体,如氨基酸和羟基酸,由于其优越的性能,如结构的可调性和良好的生物相容性,以及各种活性基团,如巯基(
‑
sh)、氨基(
‑
nh2)、羟基(
‑
oh)和羧基(
‑
cooh),为开发靶向纳米药物递送系统提供了较大的机会。近年来,基于羟基酸已经合成了许多聚酯类聚合物,新型聚羟基酸的研究为如何提高聚羟基酸的生物安全性及自身治疗能力提供了新的思路,但它们都是基于一种羟基酸反应形成聚酯,依旧存在结构和功能单一的局限,很难制备出具有多功能性的聚合物纳米载体。尤其对于生物大分子药物的装载和递送,存在结构上和功能上的不足。如申请号为cn 201810820955.x的中国发明专利,公开了一种聚阿魏酸纳米药物载体的制备及应用。通过在吡啶环境中将阿魏酸与氯化亚砜进行缩聚反应质保聚阿魏酸。该类聚合物具有良好的生物相容性和生物可降解性,适用于制备包载油溶性药物的纳米颗粒。然而该共聚物递药体系虽然改善了肿瘤药物的水溶性,但存在载药量低且释放较慢的缺陷。为了实现生物大分子药物的高效递送,并且精准探究聚合物结构功能与生物大分子药物递送效率的关系,本发明公开了一种基于氨基酸和羟基酸共聚的纳米载药体系及其制备方法和应用。
技术实现要素:
4.本发明要解决的技术问题是针对上述抗肿瘤药物递送中的问题,提供一种适用于抗肿瘤药物高效递送的纳米载药体系。本发明从机体内大量存在的氨基酸出发,利用快速
缩聚反应与羟基酸合成基于氨基酸和羟基酸共聚的新型生物相容性高、生物可降解、且具有一定的ph响应的功能性高分子,实现以ptx为代表的疏水性抗肿瘤药物的高效传递、以及其在肿瘤细胞内的可控释放。该纳米载药体系具有很好的生物安全性、生物可降解性和长循环稳定性,具备高效肿瘤靶向和高效抑制肿瘤细胞生长的特点,可有效解决纳米药物载体的溶解性差、靶向性差、响应性不好、释放速度慢和体循环稳定性较差的问题。
5.本发明的第一个目的是提供一种氨基酸
‑
羟基酸共聚物(pah)的制备方法。
6.本发明的第二个目的是提供上述方法制备得到的聚氨基酸
‑
羟基酸共聚物。
7.本发明的第三个目的是提供上述聚氨基酸
‑
羟基酸共聚物(pah)在制备抗肿瘤药物载体中的应用。
8.本发明的第四个目的是提供一种ph响应型药物输送载体的制备方法。
9.本发明的第五个目的是提供一种基于上述聚氨基酸
‑
羟基酸共聚物且具有ph响应性的纳米载药体系。
10.本发明的第六个目的是提供一种用于抗肿瘤药物递送的纳米递药体系的制备方法。
11.本发明上述目的通过以下技术方案实现:
12.一种氨基酸
‑
羟基酸共聚物的制备方法,具体包括以下步骤:
13.s1、在冰水浴条件下,将氯化亚砜滴加到溶剂中,搅拌混合均匀,得到混合溶液;
14.s2、将氨基酸或其盐和羟基酸或其盐缓慢加入到步骤s1中得到的混合溶液中,在室温条件下搅拌反应,得到氨基酸
‑
羟基酸共聚物;
15.或
16.s3、将氨基酸或其盐和羟基酸或其盐溶解到溶剂中,然后加入偶联剂和催化剂,在室温条件下进行反应,得到氨基酸
‑
羟基酸共聚物。
17.在本发明较佳的实施例中,步骤s1中所述的搅拌混合反应需在冰水浴中进行,温度过高会导致放热严重使氯化亚砜挥发且后续加入氨基酸或其盐和羟基酸或其盐时导致副产物增加,致使产物产率和纯度较低,纳米粒稳定性较差。
18.在本发明较佳的实施例中,步骤s1中所述的冰水浴的温度为0~4℃。
19.在本发明较佳的实施例中,步骤s1中所述的反应需要在无水条件下进行,水的存在会与氯化亚砜反应,使产率降低,因此,步骤s1中的溶剂选自吡啶或含有二甲基甲酰胺的二氯甲烷溶液。
20.在本发明较佳的实施例中,步骤s1中所述的氯化亚砜的滴加速度为0.2~2ml/min。氯化亚砜要缓慢滴加至用冰水浴孵育的溶剂中,以防止加入过快导致放热严重使氯化亚砜挥发且后续加入氨基酸或其盐时导致副产物增加,致使产物产率和纯度较低,纳米粒稳定性较差。
21.步骤s1中氯化亚砜与溶剂的用量比是1:15~1:20。
22.在本发明较佳的实施例中,步骤s1中所述的吡啶量不可过多,否则会导致后续处理较复杂。
23.在本发明较佳的实施例中,步骤s1中得到氯化亚砜的混合液后,将其转移到室温环境中,以待与氨基酸或其盐反应。
24.在本发明较佳的实施例中,步骤s1中所述的搅拌的时间为10~30min。
25.在本发明较佳的实施例中,步骤s2和s3中所述的氨基酸或其盐为疏水性氨基酸、疏水性氨基酸衍生物和疏水性氨基酸盐中的至少一种;进一步优选为苯丙氨酸、苯丙氨酸衍生物、苯丙氨酸盐、甲硫氨酸、甲硫氨酸衍生物、甲硫氨酸盐、缬氨酸、缬氨酸衍生物、缬氨酸盐、亮氨酸、亮氨酸衍生物、亮氨酸盐、色氨酸、色氨酸衍生物和色氨酸盐中的至少一种;更进一步优选为苯丙氨酸、苯丙氨酸衍生物和苯丙氨酸盐中的至少一种;再进一步优选为苯丙氨酸、苯丙氨酸盐酸盐和苯丙氨酸硫酸盐中的至少一种;最优选为l
‑
苯丙氨酸、d
‑
苯丙氨酸或l,d
‑
苯丙氨酸。
26.在本发明较佳的实施例中,步骤s2和s3中所述的羟基酸或其盐为脂肪族/芳香族羟基酸或其盐中的至少一种。进一步优选为水杨酸、水杨酸衍生物、水杨酸盐、阿魏酸、阿魏酸衍生物、阿魏酸盐、果酸、果酸衍生物、果酸盐、咖啡酸、咖啡酸衍生物、咖啡酸盐、香豆酸、香豆酸盐、香豆酸衍生物等羟基酸或其盐中的至少一种。更进一步优选为水杨酸、水杨酸衍生物、水杨酸钠中的至少一种;最优选为水杨酸。
27.在本发明较佳的实施例中,步骤s2中所述的氨基酸或其盐和羟基酸或其盐的总量与所述氯化亚砜的摩尔比为1:1~4;优选为1:1~2;更优选为1:1.2,所述氨基酸或其盐与羟基酸或其盐的摩尔为1~3:1~3。氯化亚砜的用量和反应温度会显著影响合成的聚合物的产率和稳定性,如果氯化亚砜加入过量,则会产生较多副产物和低聚物,致使产物产率和纯度较低,纳米粒稳定性较差。
28.在本发明较佳的实施例中,步骤s2中所述的反应的时间为0.5~12h;优选为3h。
29.在本发明较佳的实施例中,所述的氨基酸
‑
羟基酸共聚物的制备方法,在步骤s2之后还包括纯化和干燥的步骤,具体为:将步骤s2中得到的氨基酸
‑
羟基酸共聚物倒入水中搅拌,以终止反应,然后离心,除去未反应单体和溶剂,重复3~4次,真空干燥,得到氨基酸
‑
羟基酸共聚物。
30.在本发明较佳的实施例中,所述的离心的转速为3000~8000rpm;优选为5000rpm。所述的真空干燥的条件为:50~80℃真空干燥12~24h。优选为60℃真空干燥24h。
31.在本发明较佳的实施例中,步骤s2中所述的氨基酸
‑
羟基酸共聚物为聚苯丙氨酸
‑
水杨酸(ppsa),其聚合度为2~9(优选为5~6),分子量为308~1377。共聚物的聚合度和分子量会影响其自组装成纳米粒子时的粒径大小等,当共聚物ppsa的聚合度为5~6时,其自组装成纳米粒子时粒径均一、尺寸合适,能充分发挥载药纳米粒的epr效应,增强抗肿瘤效果。
32.在本发明较佳的实施例中,步骤s3中所述的偶联剂为1
‑
(3
‑
二甲氨基丙基)
‑3‑
乙基碳二亚胺盐酸盐(edc)或二环己基碳二亚胺(dcc)。
33.在本发明较佳的实施例中,步骤s3中所述的溶剂为水,二甲基甲酰胺(dmf)和二甲基亚砜(dmso)中的至少一种。
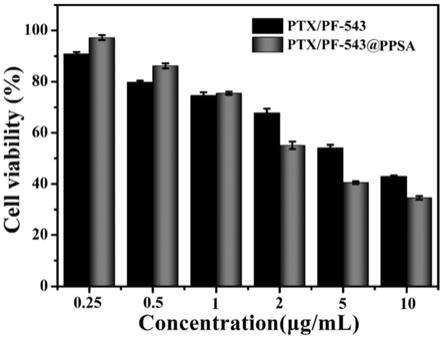
34.在本发明较佳的实施例中,步骤s3中所述的偶联剂与氨基酸或其盐和羟基酸或其盐总量的摩尔比为1.2~2:1,其中所述氨基酸或其盐与羟基酸或其盐的摩尔比为1~3:1~3。
35.在本发明较佳的实施例中,步骤s3中所述的催化剂为1
‑
羟基苯并三唑(hobt),n
‑
羟基琥珀酰亚胺(nhs)和4
‑
二甲氨基吡啶(dmap)中的至少一种。
36.在本发明较佳的实施例中,步骤s3中所述的催化剂与氨基酸或其盐和羟基酸或其
盐总量的摩尔比是1~4:1。
37.在本发明较佳的实施例中,步骤s3中所述的反应的时间为12~72h;优选为48h。
38.在本发明较佳的实施例中,所述的氨基酸共聚物的制备方法,在步骤s3之后还包括纯化和干燥的步骤,具体为:将步骤s3得到的氨基酸
‑
羟基酸共聚物加入透析袋中,然后放入水中进行透析,冻干,得到氨基酸
‑
羟基酸共聚物。
39.在本发明较佳的实施例中,所述的透析的截留分子量为3500da。所述的透析的时间优选为3d,每8h换一次水。
40.在本发明较佳的实施例中,所述的氨基酸
‑
羟基酸共聚物在制备过程需严格控制反应条件和反应比例,反应温度显著影响获得的聚合材料的性能效果,如果反应温度过高或时间过长,材料内部的功能键生成速率加快,会造成其分子量分布变宽,从而得到的产品稳定性能会下降;而反应温度过低或时间过短,造成其内部不能形成足够且有效的功能结构,进而影响载体递药效率和释药性能。
41.一种聚氨基酸
‑
羟基酸共聚物,由上述方法制备得到。
42.所述氨基酸
‑
羟基酸共聚物在制备药物载体中的应用。所述的应用的环境为体内外环境。
43.一种聚氨基酸
‑
羟基酸共聚物药物输送载体的制备方法,包括以下步骤:
44.(1)将上述氨基酸
‑
羟基酸共聚物溶于有机溶剂中,得到氨基酸
‑
羟基酸共聚物溶液;然后在搅拌条件下,将氨基酸
‑
羟基酸共聚物溶液滴加至含有稳定剂的水溶液中,使其自组装成纳米粒,得到氨基酸
‑
羟基酸共聚物药物输送载体;
45.或
46.(2)将上述氨基酸
‑
羟基酸共聚物和稳定剂分别溶解于有机溶剂中,得到氨基酸
‑
羟基酸共聚物溶液和稳定剂溶液;然后将两者混合均匀后滴加至水中,使其自组装成纳米粒,得到聚氨基酸
‑
羟基酸药物输送载体。
47.在本发明较佳的实施例中,方式(1)和(2)中所述的稳定剂为聚乙烯醇(pva),两性离子活性剂或dspe
‑
peg(二硬脂酰基磷脂酰乙醇胺
‑
聚乙二醇);优选为dspe
‑
peg
2000
。
48.在本发明较佳的实施例中,所述的两性离子活性剂优选为羧基甜菜碱或磺基甜菜碱。
49.在本发明较佳的实施例中,方式(1)和(2)中所述的稳定剂的用量为占氨基酸
‑
羟基酸共聚物质量的0~75%(不包括0);优选为占氨基酸
‑
羟基酸共聚物质量的25~50%;更优选为占共聚物质量的50%。
50.在本发明较佳的实施例中,方式(1)和(2)中所述的有机溶剂为二甲基亚砜(dmso)、n,n
‑
二甲基甲酰胺(dmf)和四氢呋喃(thf)中的一种或多种;优选为二甲基亚砜(dmso)。
51.在本发明较佳的实施例中,方式(1)和(2)中所述的氨基酸
‑
羟基酸共聚物溶液的浓度为5~50mg/ml;优选为10~50mg/ml;更优选为10~20mg/ml。
52.在本发明较佳的实施例中,方式(1)中所述稳定剂的水溶液和(2)中所述的稳定剂溶液的浓度均为5~50mg/ml;优选为10~50mg/ml;更优选为10~20mg/ml。
53.在本发明较佳的实施例中,所述的氨基酸
‑
羟基酸共聚物药物输送载体的制备方法,还包括将得到的氨基酸
‑
羟基酸共聚物药物输送载体去除溶剂的步骤,具体为:将自组
装成纳米粒置于超滤管中离心,重复3次以上,使有机溶剂的含量在千分之一以下,得到去除溶剂后的聚氨基酸
‑
羟基酸药物输送载体。所述超滤管为截留分子量为100kda的超滤管。所述的离心的条件为:2000~3000rpm离心8~15min;优选为:3000rpm离心10min。
54.一种聚氨基酸
‑
羟基酸共聚物的纳米载药体系,包括上述方法制得的聚氨基酸
‑
羟基酸共聚物以及抗肿瘤药物和/或蛋白酶类抑制剂。
55.在本发明中,对所载的抗肿瘤药物没有特殊限制,所述的抗肿瘤药物包括亲水性药物和疏水性药物;所述亲水性药物包括但不限于盐酸阿霉素、盐酸吉西他滨、盐酸伊立替康、氟尿嘧啶或香菇多糖等药物;所述疏水性药物包括但不限于紫杉醇(ptx)、多西紫杉醇、甲氨蝶呤、喜树碱、阿霉素、姜黄素等药物。
56.在本发明中,对所载的蛋白酶类抑制剂没有特殊限制,所述的蛋白酶类抑制剂包括共价cdk7抑制剂(thz1)、胆固醇转脂酶抑制剂(avasimibe)、鞘氨醇激酶抑制剂(pf
‑
543)、硼替佐米(bortezomib,1)、钙蛋白酶抑制剂、蛋白酶体抑制剂(lactacystin)等蛋白酶类抑制剂。
57.一种聚氨基酸
‑
羟基酸共聚物的纳米载药体系的制备方法,具体包括以下步骤:
58.(i)将上述氨基酸
‑
羟基酸共聚物、抗肿瘤药物、稳定剂分别溶解到有机溶剂中,得到氨基酸
‑
羟基酸共聚物溶液、抗肿瘤药物溶液、稳定剂溶液;然后将几种溶液混合均匀后滴加至水中,使其自组装成载药纳米粒,得到聚氨基酸
‑
羟基酸纳米载药体系;
59.或
60.(ii)将上述氨基酸
‑
羟基酸共聚物、抗肿瘤药物分别溶解到有机溶剂中,得到聚氨基酸
‑
羟基酸溶液、抗肿瘤药物溶液;然后将氨基酸
‑
羟基酸共聚物溶液和抗肿瘤药物溶液缓慢滴加到含有稳定剂的水溶液中,使其自组装成纳米粒,得到聚氨基酸
‑
羟基酸纳米载药体系。
61.在本发明较佳的实施例中,方式(i)和(ii)中所述的稳定剂为聚乙烯醇(pva),两性离子活性剂或dspe
‑
peg(二硬脂酰基磷脂酰乙醇胺
‑
聚乙二醇);优选为dspe
‑
peg
2000
。
62.在本发明较佳的实施例中,所述的两性离子活性剂优选为羧基甜菜碱或磺基甜菜碱。
63.在本发明较佳的实施例中,方式(i)和(ii)中所述的有机溶剂为二甲基亚砜(dmso)、n,n
‑
二甲基甲酰胺(dmf)和四氢呋喃(thf)中的一种或多种;优选为二甲基亚砜(dmso)。
64.在本发明较佳的实施例中,方式(i)和(ii)中所述的氨基酸
‑
羟基酸共聚物溶液的浓度为5~50mg/ml;优选为10~50mg/ml;更优选为10~20mg/ml。
65.在本发明较佳的实施例中,方式(i)和(ii)中所述的抗肿瘤药物溶液的浓度为10~50mg/ml;更优选为10~20mg/ml。
66.在本发明较佳的实施例中,方式(i)和(ii)中所述抗肿瘤药物可以替换成蛋白酶类抑制剂,所得蛋白酶类抑制剂溶液的浓度为10~50mg/ml;更优选为10~20mg/ml。
67.在本发明较佳的实施例中,方式(i)和(ii)中所述抗肿瘤药物还可以与蛋白酶类抑制剂一起使用制备纳米载药体系,所得抗肿瘤药物溶液和蛋白酶类抑制剂溶液的浓度均为10~50mg/ml;更优选为10~20mg/ml。
68.在本发明较佳的实施例中,方式(i)和(ii)中所述的稳定剂溶液和方式(ii)中所
述稳定剂的水溶液浓度均为10~50mg/ml;更优选为10~20mg/ml。
69.在本发明较佳的实施例中,方式(i)和(ii)中所述的氨基酸
‑
羟基酸共聚物与抗肿瘤药物的质量比为1:0.05~0.50;优选为1:0.1~0.5,更优选为1:0.30。氨基酸
‑
羟基酸共聚物和抗肿瘤药物的质量比过大,则载药量较低,所成纳米粒粒子数较少,过小则包封率较小,造成药物的浪费,并且所得纳米粒不稳定,易沉淀。
70.在本发明较佳的实施例中,当所述抗肿瘤药物替换成蛋白酶类抑制剂时,方式(i)和(ii)中所述的氨基酸
‑
羟基酸共聚物与蛋白酶类抑制剂的质量比为1:0.05~0.50;优选为1:0.1~0.5,,更优选为1:0.3。
71.在本发明较佳的实施例中,当同时含有抗肿瘤药物和蛋白酶类抑制剂时,方式(i)和(ii)中所述的氨基酸
‑
羟基酸共聚物与抗肿瘤药物、蛋白酶类抑制剂的质量比为1:0.05~0.50:0.05~0.5;优选为1:0.1~0.5:0.1~0.5。
72.在本发明较佳的实施例中,方式(i)和(ii)中所述的稳定剂的用量占氨基酸
‑
羟基酸共聚物质量的0~75%(不包括0);优选为占氨基酸
‑
羟基酸共聚物质量的25~50%;更优选为占氨基酸
‑
羟基酸共聚物质量的50%。
73.在本发明较佳的实施例中,所述的氨基酸
‑
羟基酸共聚物的纳米载药体系,还包括将得到的氨基酸
‑
羟基酸共聚物的纳米载药体系进行纯化的步骤,具体为:将自组装成纳米粒置于超滤管中,离心,重复3次以上,以除去未包封的抗肿瘤药物以及使有机溶剂的含量在千分之一以下,得到纯化后的氨基酸
‑
羟基酸共聚物的纳米载药体系。所述超滤管为截留分子量为100kda的超滤管。所述的离心的条件为:2000~3000rpm离心8~15min;优选为:3000rpm离心10min。
74.在本发明中,所述的聚氨基酸纳米载药体系(载药纳米粒)的粒径为50~180nm,拥有较高的比表面积,载药量高,是优良的载药体系,可增强药物的疗效,而且该载药纳米粒子能够利用epr效应增强药物在肿瘤部位的靶向性。
75.本发明中的聚氨基酸
‑
羟基酸纳米载药体系所需原料易得,制备工艺纯熟,易于操作,不需要昂贵的仪器,且制备的纳米复合物尺寸适中、生物相容性好,不仅实现了疏水药物的可控载药,改善了药物的溶解性,极大的提高了疏水药物的可利用性,还可以大大提高纳米粒在血液中的循环时间,从而提高肿瘤部位的药物聚积,提高治疗效果。
76.本发明相对于现有技术具有如下的优点及效果:
77.(1)本发明针对抗肿瘤药物的递送效率较低及聚氨基酸
‑
羟基酸合成条件及工艺相对苛刻的问题,提供一种能够快速而简便地制备且适用于抗肿瘤药物递送的纳米载药体系。本发明以苯丙氨酸和水杨酸为例,即以苯丙氨酸,水杨酸和氯化亚砜(socl2)为原料,通过一步缩聚法在吡啶(碱性环境中)快速而简便地发生缩聚反应来制备聚苯丙氨酸
‑
羟基酸(ppsa)。该共聚物具有较好的生物相容性,制备而成的纳米粒载体具有载药量高且尺寸稳定,能够通过肿瘤组织的增强渗透滞留效应(epr)较好地聚集在肿瘤部位,在提高化疗药物的生物利用度和降低化疗药物对正常组织的毒性的同时增强了纳米药物载体对肿瘤细胞生长抑制的特点。与开环聚合制备聚氨基酸
‑
羟基酸相比,本发明具有反应周期短、条件温和、重复性好等优势,具有良好的应用前景和发展空间。此外,该纳米载体本身具有一定的抗炎和抗肿瘤作用,为癌症及其他疾病的有效治疗开拓了一种新的途径。
78.(2)本发明材料来源于生物相容性好的氨基酸和羟基酸,安全性有保障,其制备方
法简单,聚苯丙氨酸
‑
羟基酸抗肿瘤递药体系改善了药物的溶解性,极大的提高了疏水药物的可利用性,还可以大大提高纳米粒在血液中的循环时间,从而提高肿瘤部位的药物聚积,并且载药量高,有一定的酸响应性,绿色安全,易于实现,同时降低了化疗药物对正常组织的毒性,拓宽了其在抗肿瘤方面的应用范围。
79.(3)本发明从机体内大量存在的苯丙氨酸出发,合成基于苯丙氨酸的新型生物相容性高、生物可降解的高分子,同时改善了药物的溶解性,极大的提高了疏水药物的可利用性,还可以大大提高纳米粒在血液中的循环时间,从而提高肿瘤部位的药物聚积。另外,肿瘤组织具有不同于正常组织的微环境,与正常细胞相比,肿瘤组织呈弱酸性,酯键和酰胺键在肿瘤细胞质中较低ph下会加快药物释放,从而引发药物的释放至肿瘤细胞,并且随后可高效诱导肿瘤细胞凋亡。同时,本发明使用的具有较好疏水性的l
‑
苯丙氨酸可将肿瘤药物分子直接导入癌瘤区,大大增强了靶向性,同时水杨酸具有一定的抗炎和抗肿瘤作用,这样既可以抑制癌瘤生长,又可以降低药物的毒副作用。
80.(4)本发明从机体内大量存在的苯丙氨酸出发与水杨酸通过一步缩聚法快速简便地合成基于苯丙氨酸和水杨酸的新型共聚物,该共聚物具有生物相容性高、生物可降解的特点,可实现以ptx为代表的疏水性抗肿瘤药物的高效传递、以及其在肿瘤细胞内的释放。该纳米载药体系具有很好的生物安全性、生物可降解性和长循环稳定性,具备高效肿瘤靶向和高效抑制肿瘤细胞生长的特点,可有效解决纳米药物载体的溶解性差、靶向性差和体循环稳定性较差的问题。
81.(5)不同的共聚物结构对于载药和递送性能有不同程度的影响,本发明所制备的纳米递药体系中,充分将聚合物的结构、性能和亲疏水性等因素考虑在内,通过调整材料的物理化学性质,制备出粒径合适、亲水性适宜、生物可降解聚苯丙氨酸
‑
羟基酸聚合物,再与抗癌药物复合形成纳米结构,可以实现抗癌药物的高效负载和可控释放,表现出良好的生物安全性,体内循环稳定性好,同时由于本发明载药纳米颗粒具有一定的酸响应性,其在肿瘤细胞较低ph环境中,可导致聚合物纳米粒的快速崩解和包载药物的迅速释放,增强了肿瘤细胞对于抗癌药物的利用,从而在增强肿瘤细胞杀伤作用的同时,达到降低化疗药物对正常组织毒性的目的。
附图说明
82.图1为实施例1制备的聚苯丙氨酸
‑
水杨酸的核磁氢谱解析图。
83.图2为实施例1制备的聚苯丙氨酸
‑
水杨酸的红外光谱解析图。
84.图3为通过飞行质谱对实施例1制备的聚苯丙氨酸
‑
水杨酸的分子量表征图。
85.图4为实施例2制备的聚苯丙氨酸
‑
水杨酸(药物输送载体)的纳米载体粒径分布及透射电镜图。
86.图5为实施例2制备的聚苯丙氨酸
‑
水杨酸(药物输送载体)的生物相容性表征图;其中,a为材料对4t1细胞的毒性实验结果;b溶血实验。
87.图6为实施例4制备的纳米药物载体与游离药物对4t1细胞的杀伤作用。
88.图7为实施例4制备的纳米药物载体对4t1细胞的迁移的抑制作用;其中a为具有代表性的不同组4t1细胞迁移的图像,b为对不同组细胞迁移情况的定量分析。
89.图8为实施例4制备得到的载药纳米粒的ptx/pf543@ppsa作用于4t1细胞的凋亡结
果图;其中a为4t1细胞的流式凋亡结果图,b为对不同组细胞凋亡的定量分析。
90.图9为实施例4制备得到的载药纳米粒ptx/pf543@ppsa对4t1肿瘤小鼠的抑瘤效果图。
91.图10为用实施例4制备得到的载药纳米粒ptx/pf543@ppsa处理小鼠后对小鼠生理状况的影响
具体实施方式
92.下面结合实施例对本发明作进一步详细的描述,但本发明的实施方式不限于此。除非特别说明,本发明采用的试剂、方法和设备为本技术领域常规试剂、方法和设备。下列实施例中未注明具体实验条件的试验方法,通常按照常规实验条件或按照制造厂所建议的实验条件。除非特别说明,本发明所用试剂和原材料均可通过市售获得。
93.本发明提供了一种聚氨基酸
‑
羟基酸的制备方法(选取苯丙氨酸和水杨酸为例),包括以下步骤:
94.(1)在冰水浴条件下,将一定量的氯化亚砜缓慢滴加(滴加速度0.2~2ml/min)于盛有一定量的无水吡啶反应瓶中搅拌10~30min,得到混合溶液;其中,氯化亚砜与无水吡啶的体积比为1:20~30;
95.(2)在一定温度下,将一定量的疏水性氨基酸或其盐(本发明中以苯丙氨酸为例)和疏水性羟基酸(本发明以水杨酸为例)缓慢加入到上述混合溶液,继续搅拌一定时间,得到聚苯丙氨酸
‑
水杨酸(ppsa);其中,氯化亚砜与苯丙氨酸(或其盐)和水杨酸(或其盐)的摩尔之和的比为1~4:1(优选为1~2:1),更优选为1.2:1,其中苯丙氨酸与水杨酸的比为1~3:1~3),反应温度为室温,反应时间为0.5~12h(优选为3h),此条件下获得的聚苯丙氨酸
‑
水杨酸(ppsa)的产率较高、稳定性高;
96.(3)将反应所得的ppsa倒入水中搅拌,以终止反应,之后将产物置于离心管中3000~8000rpm条件下离心,除去未反应单体和吡啶,重复3~4次后在50~80℃真空干燥12~24h,得到浅黄色粉末状聚合物。
97.除上述方法外,还可以将氨基酸或其盐和水杨酸(或其盐)与偶联剂和催化剂反应得到氨基酸
‑
羟基酸聚合物(此方法合成材料周期相对较长些,产率相对较低些),具体为:
98.(i)在一定温度下,将一定量的疏水性氨基酸或其盐(以苯丙氨酸为例)疏水性羟基酸(以水杨酸为例)(加入到溶剂(水,dmf或dmso)中,然后加入偶联剂edc(或dcc)和催化剂hobt(nhs或dmap)进行反应;其中,偶联剂与氨基酸和羟基酸的摩尔之和的比为1.2~2:1;催化剂与氨基酸和羟基酸的摩尔比是1~4:1;反应温度为室温,反应时间为12~72h(优选为48h)。
99.(ii)反应结束后,将步骤(i)获得的产物加入透析袋(截留分子量为3500da)中,之后放入水中透析3d,每8h换一次水,冻干,得到氨基酸
‑
羟基酸共聚物。
100.实施例1聚苯丙氨酸
‑
羟基酸的制备(本实施例中的氨基酸和羟基酸分别为l
‑
苯丙氨酸和水杨酸)
101.1、一种共聚物ppsa的制备方法,包括以下步骤:
102.(1)在冰水浴条件下,将氯化亚砜缓慢滴加(滴加速度0.2~2ml/min)于盛有无水吡啶的反应瓶中搅拌10~30min,得到混合溶液;
103.(2)将苯丙氨酸和水杨酸缓慢加入到上述混合溶液,继续搅拌10~30min,得到聚苯丙氨酸
‑
水杨酸(ppsa);其中,氯化亚砜与苯丙氨酸和水杨酸的摩尔之和的比分别为1:1、1.2:1、2:1、3:1和4:1,其中苯丙氨酸和水杨酸的摩尔比为1~3:1~3,反应温度为室温,反应3h。
104.(3)在搅拌情况下,将反应所得的ppsa滴加至水中,以终止反应,之后将产物置于离心管中5000rpm条件下离心,除去未反应单体和吡啶,重复3~4次后在60℃真空干燥24h,得到一系列浅黄色粉末状共聚物ppsa。
105.2、结果
106.本实施例利用400m超导核磁共振波谱仪(ascend tm 400)和布鲁克红外光谱仪(vertex70)表征实施例1制备的共聚物ppsa的结构。
107.结果如图1所示,图1的核磁氢谱图中,在约8.25ppm处的峰对应聚苯丙氨酸
‑
羟基酸酰胺键上的
‑
nh的峰;在7.25
‑
7.32ppm左右的峰则是ppsa的苯环上氢的峰,在4.5ppm左右的峰则是苯丙氨酸上的
‑
ch上的氢,而3.0ppm左右的峰则对应于苯丙氨酸上亚甲基的氢,核磁氢谱表明该共聚物已被成功制备。进一步的结构信息则由红外表征,见图2红外测试结果(图中仅以氯化亚砜与苯丙氨酸和水杨酸的比为1.2:0.5:0.5)为代表进行表征,图中未显示其他比例的结果,但其他结果与此图相似),3300cm
‑1左右的峰为共聚物酰胺键中n
‑
h的伸缩振动,3000
‑
3100cm
‑1、1455cm
‑1以及700cm
‑1为ppsa中苯环的特征吸收峰;而1250cm
‑1为酰胺键上c
‑
n伸缩振动;1641cm
‑1为酰胺ⅰ吸收带。以上结果表明共聚物ppsa被成功合成。
108.另外,当氯化亚砜与苯丙氨酸和水杨酸的摩尔之和的比为1~2:1时,更优选为1.2:1时(其中苯丙氨酸和水杨酸的摩尔1:1),所得共聚物ppsa结构稳定、生物相容性更好,能包载不同亲疏水性的药物,所制备的共聚物ppsa具有粒径小、稳定性高的特征,在作为肿瘤药物传递载体方面具有良好应用前景。
109.结果如图3所示,本实施例制备共聚物ppsa的聚合度是2~9,分子量范围在308~1377。共聚物的聚合度和分子量会影响其自组装成纳米粒子时的粒径大小等。当共聚物ppsa的聚合度是5~6时,其自组装成纳米粒子时粒径均一、尺寸合适,能充分发挥载药纳米粒的epr效应,增强抗肿瘤效果。
110.本实施例共聚物ppsa在制备过程需严格控制反应条件,氯化亚砜需在冰水浴条件下缓慢加入吡啶溶液中,温度过高会使氯化亚砜蒸出且有副产物产生,磁力搅拌一定时间使其充分混匀。在快速搅拌条件下将苯丙氨酸和水杨酸混合后缓慢加入含氯化亚砜的吡啶溶液,以防止成环或者“夹生”,致使产物产率和纯度较低,纳米粒稳定性较差。
111.实施例2聚苯丙氨酸
‑
羟基酸纳米粒的制备
112.1、聚苯丙氨酸
‑
羟基酸纳米粒的制备过程包括以下步骤:
113.(1)将实施例1制备得到的不同的共聚物ppsa(本实施例选用氯化亚砜与苯丙氨酸和水杨酸的比为1.2:0.5:0.5制备得到的ppsa)和表面稳定剂dspe
‑
peg
2000
分别溶于二甲基亚砜(dmso)中,分别配成10mg/ml的溶液备用;
114.(2)将上述配制的ppsa与表面稳定剂dspe
‑
peg
2000
溶液按照一定比例混合后在1000rpm转速下缓慢滴加至水溶液中,该复合物在水溶液中通过纳米沉淀法自组装形成纳米体系;其中,dspe
‑
peg
2000
的质量为ppsa质量的50%;
115.(3)将所得纳米粒溶液置于截留分子量(mwco=100kda)的超滤管中,3000rpm超滤
3次,每次10min,使dmso含量在千分之一以下,最后得到ppsa纳米粒。
116.2、物理性质表征和细毒性实验
117.(1)利用纳米粒径仪和透射电镜对其物理性质进行表征。
118.(2)对该纳米粒子进行生物相容性实验,具体步骤为:
119.细毒性实验:将状态良好的4t1细胞(北京北纳创联生物技术研究院)接种在96孔板中(每孔5,000个细胞),并培养24小时。然后用不同浓度(5,10,50,100,200μg/ml)的ppsa 处理细胞24h,往每孔添加20μl mtt,再孵育4h,之后弃去培养基,向每个孔中添加180μl dmso。随后,将板轻轻摇动15分钟以溶解甲瓒,并在490nm测吸光度。
120.溶血实验:根据已报道的方案并稍作修改对聚苯丙氨酸
‑
羟基酸的溶血活性进行评估。简而言之,从sd大鼠(中山大学实验动物中心,220~280g)眼眶取新鲜血液并用生理盐水洗涤,通过离心法从血液中分离出红细胞(rbc)后,用生理盐水配成一定浓度的红细胞悬液之后加入不同浓度的ppsa溶液(5,10,50,100,200μg/ml)并温和地涡旋混合;将混合物置于37℃空气摇床震荡3h,然后将样品离心并转移至96孔板;使用酶标仪在540nm处测量上清液中的游离血红蛋白的吸光度,其中,生理盐水和超纯水分别作为阴性和阳性对照。
121.3、结果
122.(1)利用纳米粒经仪和透射电镜对其物理性质进行表征。结果如图4所示:所制得的纳米粒子粒径90nm,呈类圆形,大小相对均一,在pbs和complete medium中具有较好的稳定性。
123.(2)细毒性实验和溶血实验结果如图5所示:图5a表明该纳米粒子具有较好的细胞相容性,所制备的纳米粒用作药物载体具有较好的应用前景;图5b表明所制备的纳米粒子具有较好的血液相容性,能够用于动物体内。
124.实施例3聚苯丙氨酸
‑
羟基酸纳米粒的制备
125.方法同实施例2步骤1,不同之处在于:将ppsa溶解于n,n
‑
二甲基甲酰胺(dmf)中,并控制ppsa的浓度分别为5mg/ml、10mg/ml、50mg/ml,最后得到的ppsa纳米粒。
126.实施例4聚苯丙氨酸
‑
羟基酸纳米载药体系的制备
127.1、聚苯丙氨酸纳米载药体系的制备过程包括以下步骤:
128.(1)本实施例以抗肿瘤药物紫杉醇(ptx)和蛋白抑制剂pf
‑
543为代表进行制备,将ptx、pf
‑
543、表面稳定剂dspe
‑
peg
2000
,实施例1制备得到的不同的ppsa分别溶于dmso中,分别配成相同浓度的溶液(10mg/ml)备用;
129.(2)将等浓度四种溶液,按照共聚物ppsa和抗肿瘤药物以及蛋白抑制剂pf
‑
543的质量比为10:1:2,10:1:3,10:2:1,10:2:2,10:2:3,10:3:2以及10:3:3的不同比例混合后,按dspe
‑
peg
2000
占共聚物ppsa质量的0~75%进行混合,在1000rpm转速下缓慢滴加至水溶液中,该复合物在水溶液中通过纳米沉淀法自组装形成纳米体系;其中,本案例较佳dspe
‑
peg
2000
质量为ppsa质量的50%;
130.(3)将所得纳米粒溶液置于截留分子量(mwco=100kda)的超滤管中,3000rpm超滤3次,每次10min,以除去未包封的ptx和pf
‑
543并使dmso含量在千分之一以下,最后得到ptx/pf543@ppsa nps载药纳米粒。
131.2、结果
132.在共聚物ppsa和其他条件相同的条件下,以共聚物ppsa与ptx以及pf
‑
543的质量
al.pursuing specific chemotherapy of orthotopic breast cancer with lung metastasis from docking nanoparticles driven by bioinspired exosomes[j].nano letter,2019,19(5).),通过在balb/c小鼠的第四对乳房垫下注射4t1细胞(约2
×
106)建立原位乳腺癌荷瘤小鼠模型。动物实验根据中山大学机构动物护理和使用委员会(iacuc)批准的协议(证书编号,sysu
‑
iacuc
‑
2019
‑
000152)进行。当小鼠的肿瘤体积达到约100mm3时,将它们随机分为四组(n=5),并隔日通过尾部静脉内注射生理盐水,ptx/pf543,ppsa,ptx/pf543@ppsa nps治疗21天。之后将小鼠处死,进行组织切片染色、血生化指标检测、免疫荧光染色等处理,来对其进行体内抗肿瘤作用评价。
[0143]
体内抑瘤效果如图9所示:tunel切片图表明载药纳米粒子具有更好的抑瘤效果,小鼠治疗21天后将对小鼠进行血生化检测如图10所示,载药纳米粒子可降低游离药物的毒性。本发明制备的纳米载药体系在肿瘤治疗领域有一定的应用潜力。
[0144]
上述结果说明,本发明将具有生理活性的疏水性苯丙氨酸和水杨酸通过一步法快速简便地在溶有氯化亚砜的吡啶溶剂中反应制备聚苯丙氨酸
‑
水杨酸。该共聚物具有较好的疏水性,在提高载药量和延长血液循环时间的同时,可将抗癌药物分子和蛋白酶类抑制剂通过epr效应导入癌症部位,达到既可以快速抑制癌瘤生长,又可以降低药物毒副作用的目的。此外,该纳米载体本身具有一定的抗炎和抗肿瘤作用,为癌症及其他疾病的有效治疗开拓了一种新的途径。
[0145]
上述实施例中,所述的抗肿瘤药物除可以选择紫杉醇外,也可以选择喜树碱、盐酸阿霉素、多西紫杉醇、盐酸吉西他滨、盐酸伊立替康、氟尿嘧啶或香菇多糖、姜黄素等抗肿瘤药物,所述的蛋白酶类抑制剂除了鞘氨醇激酶抑制剂(pf
‑
543)外,也可以选择包括共价cdk7抑制剂(thz1)、胆固醇转脂酶抑制剂(avasimibe)、硼替佐米(bortezomib,1)、钙蛋白酶抑制剂、蛋白酶体抑制剂(lactacystin)等,亦得到相同或相近的结果。实际应用中,可根据具体的癌症类型选择相应的抗肿瘤药物和蛋白酶类抑制剂与聚氨基酸
‑
羟基酸按照本发明的方法合成纳米递药体系,增强抗肿瘤药物的治疗效果的有效性、可控性和安全性。
[0146]
以上具体实施方式为便于理解本发明而说明的较佳实施例,但本发明并不局限于上述实施例,即不意味着本发明必须依赖上述实施例才能实施。所属技术领域的技术人员应该明了,对本发明的任何改进,对本发明所选用原料的等效替换及辅助成分的添加、具体方式的选择等,均落在本发明的保护范围和公开范围之内。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1