一种基于假attP位点自发定向整合的通用型微环DNA表达载体及其构建方法与应用
一种基于假attp位点自发定向整合的通用型微环dna表达载体及其构建方法与应用
技术领域
1.本发明属于基因工程技术领域,具体涉及一种位点特异性整合的微环dna表达载体及其重组母质粒,特别涉及一种基于假attp位点自发定向整合的通用型微环dna表达载体及其构建方法与应用。
背景技术:
2.重组dna技术的兴起和推广,使得在实验室条件下人工构建或修饰dna分子成为了可能,这也推动了基因治疗、转基因动物研制和基因修饰等领域的发展。然而,研究者在高等动物细胞和活体基因组中进行有效的可控性基因修饰的能力十分有限。为了将外源基因整合进基因组,目前依靠的仍然是转染dna的随机整合,不仅成本高、效率低,而且由于整合位点的随机性易引起插入突变或外源基因表达沉默。
3.近年来,链霉菌噬菌体φc31整合酶能够催化链霉菌基因组中的attb位点和噬菌体基因组attp位点之间的同源重组,引起科学家的重视。由于φc31整合酶介导的重组具有单向整合、位点特异、无需外界化学能源和辅助因子、胜任大片段基因有效整合、可在多种高等动植物细胞中发挥作用及外源基因可长期高效表达等特点,故成为继cre重组酶和flp重组酶之后的又一种基因修饰的有力工具,已经越来越多地被用于基因治疗、转基因动物研制等方面,并衍生出微环dna技术。
4.φc31整合酶识别的attb位点最小为34bp,attp位点最小为39bp,重组以两者间一段3bp重叠序列“ttg”为枢纽,将噬菌体基因组整合到细菌基因组中的attb特异位点,并形成杂合位点attl和attr。其中,attb位点的34bp核心序列为gtgccagggcgtgcccttgggctccccgggcgcg,attp位点的39bp核心序列为ccccaactggggtaacctttgagttctctcagttggggg。
5.研究表明,在人、小鼠、大鼠、果蝇、牛等真核生物基因组中存在着“假attp位点”,这些位点的序列与噬菌体attp位点的序列有显著的相似性,同样可以由φc31整合酶介导发生位点特异性整合反应。此外,在真核细胞中,φc31整合酶介导的整合多发生于转录活跃区,故有利于外源基因的表达,这也可能是整合酶介导的整合与随机整合相比普遍有较长时间高水平表达的原因。而且,整合位点不偏好于转录起始位点,sivalingam等发现,超过70%的整合位点位于转录起始位点50kb以外。因此,从该方面来看,φc31整合酶介导的整合潜在的安全隐患较随机插入和病毒载体系统要小得多。
6.尽管传统的质粒载体和病毒载体是常被用于整合到宿主细胞染色体上的载体类型,但在实际应用中仍有一些问题难以克服。其中,病毒载体存在插入突变的风险和免疫原性,会带来诱癌风险并可能引发宿主的免疫反应,而质粒载体的随机整合同样会引起插入突变或导致外源基因的沉默。更重要地是,传统质粒载体中含有细菌复制序列、抗性基因、未甲基化的cpg基序和一些可能隐藏的表达信号,这些序列在质粒复制时是必需的,但在具体应用中却可能引起严重的生物安全性问题。而微环dna是一种环状的、不包含细菌质粒骨架成分(如:抗生素抗性基因和细菌复制序列)的表达载体,在生物安全性上明显优于传统
的质粒载体和病毒载体。但是,由于微环dna无法整合到宿主细胞染色体上,故只能在静止细胞中短暂表达所携带的外源基因。由此可见,目前仍然缺乏安全、可靠、具有整合功能的表达载体系统。
技术实现要素:
7.本发明解决的问题在于提供一种基于假attp位点自发定向整合的通用型微环dna表达载体及其重组母质粒的构建方法和应用,克服现有载体系统进行外源基因整合方面存在的缺陷,为外源基因整合入宿主细胞染色体提供可自发定向整合假attp位点且无细菌质粒骨架成分的微环dna表达载体,提高外源基因整合的安全性和可靠性。
8.根据本发明第一方面,本发明提供如下技术方案:
9.一种基于假attp位点自发定向整合的通用型微环dna表达载体,该微环dna表达载体由目的基因、ppap序列和微环dna质粒构成,通过在所述微环dna质粒的多克隆位点插入所述ppap序列和目的基因形成重组母质粒,再将所述重组母质粒在工程菌内发生分子内位点特异性重组而形成所述微环dna表达载体,其中所述ppap序列包括依次连接的polya加尾信号、启动子、反向attb序列、φc31整合酶基因和终止密码子,所述反向attb序列的核苷酸序列如seq id no.3所示,所述的φc31整合酶基因的核苷酸序列如seq id no.4所示,所述终止密码子的核苷酸序列为tga,所述目的基因为需要整合入宿主细胞染色体的外源基因片段。
10.优选地,所述微环dna质粒为pmc.cmv
‑
mcs
‑
sv40polya质粒或pmc.ef1α
‑
mcs
‑
sv40polya质粒。
11.优选地,所述polya加尾信号为牛生长激素多聚核苷酸bpa或sv40加尾信号;所述启动子为巨细胞病毒cmv启动子、劳斯肉瘤病毒rsv启动子、泛素ubc启动子或延长因子ef1α启动子。
12.本发明采用的polya加尾信号可以为bpa、sv40等加尾信号之一,sv40加尾信号只是本发明构建的ppap序列中的更优选polya加尾信号,本发明可以根据表达宿主细胞的不同、ppap序列插入的微环dna空质粒的不同等条件选择不同的polya加尾信号构建ppap序列。
13.本发明采用的启动子可以为cmv、rsv、ubc和ef1α等真核表达启动子之一,cmv启动子只是本发明构建的ppap序列中的更优选启动子,本发明可以根据表达宿主细胞的不同、ppap序列插入的微环dna空质粒的不同等条件选择不同的启动子构建ppap序列。
14.更优选地,所述polya加尾信号为sv40加尾信号,所述sv40加尾信号核苷酸序列如seq id no.1所示;所述启动子为巨细胞病毒cmv启动子,所述巨细胞病毒cmv启动子核苷酸序列如seq.id.no.2所示。
15.当所述polya加尾信号为sv40加尾信号、所述启动子为巨细胞病毒cmv启动子时,所述ppap序列的完整核苷酸序列如seq id no.5所示。
16.根据本发明第二方面,本发明提供如下技术方案:
17.一种上述的基于假attp位点自发定向整合的通用型微环dna表达载体的构建方法,包括如下步骤:
18.(1)采用全基因序列合成的方式合成dna片段,该dna片段包括依次连接的bstbi酶
切位点、所述ppap序列和bamhi酶切位点;
19.(2)采用bstbi内切酶和bamhi内切酶对步骤(1)所得的dna片段进行双酶切,并回收酶切后的含所述ppap序列的dna片段;
20.(3)采用bstbi内切酶和bamhi内切酶对所述微环dna质粒进行双酶切,并回收酶切后的线性质粒;
21.(4)采用dna连接酶连接步骤(2)双酶切后回收的所述dna片段和步骤(3)双酶切后回收的所述线性质粒,获得含ppap序列的微环dna重组质粒,该pmc.ef1α
‑
mcs
‑
ppap
‑
sv40polya质粒即为基于假attp位点自发定向整合的通用型微环dna表达载体的重组母质粒;
22.(5)将所述目的基因(需要整合入宿主细胞染色体的外源基因片段)插入到步骤(4)所得的微环dna重组质粒的多克隆位点并进行扩增,其中,所述的外源基因片段可采用全基因序列合成或pcr克隆等方法获得,本发明可根据实际具体情况选择最优的方法获得需要整合入宿主细胞染色体的外源基因片段,所述的pmc.ef1α
‑
mcs
‑
ppap
‑
sv40polya质粒的多克隆位点(mcs)包括的限制性内切酶识别位点为:xbai、nhei和ecori;
23.(6)将步骤(5)所得的携带需要整合入宿主细胞染色体的外源基因片段的微环dna重组母质粒转化工程菌,按照mc
‑
easy
tm minicircle dna production kit操作说明书,扩增和诱导转化后的zycy10p3s2t工程菌,然后采用常规质粒提纯试剂盒提纯由工程菌生产出的微环dna,该微环dna即为基于假attp位点自发定向整合的通用型微环dna表达载体,所述微环dna为具有attr位点、延长因子ef1α启动子、目的基因、ppap序列和polya加尾信号的环状dna。
24.优选地,所述工程菌为zycy10p3s2t工程菌。
25.根据本发明第三方面,本发明提供如下技术方案:
26.一种上述的基于假attp位点自发定向整合的通用型微环dna表达载体的应用,包括将所述微环dna表达载体转染宿主细胞。
27.优选地,所述转染宿主细胞的转染方法为化学转染方法或物理转染方法。
28.优选地,所述宿主细胞为真核细胞。
29.综上所述,由于采用了上述技术方案,本发明的有益效果如下:
30.1、本发明提供的基于假attp位点自发定向整合的通用型微环dna表达载体,在进行细胞转染后,在φc31整合酶介导下,能够位点特异性地整合到真核细胞基因组内假attp位点,其分布偏向于基因间或基因内含子内的转录活跃区域,避免了质粒随机插入引起的基因表达沉默和其他安全隐患,从而使整合入宿主细胞染色体的外源基因片段安全持续高效表达。
31.2、本发明提供的基于假attp位点自发定向整合的通用型微环dna表达载体,不包含传统质粒载体中所含有的细菌复制序列、抗性基因、未甲基化的cpg基序和一些可能隐藏的表达信号,故在具体应用中具有更高的生物安全性。
32.3、本发明提供的基于假attp位点自发定向整合的通用型微环dna表达载体,使传统的微环dna表达载体具有了自发定向整合到真核细胞基因组内假attp位点的功能,解决了表达载体整合位点、微环dna无法持续高效表达外源基因片段和质粒载体生物安全性较低等问题,可广泛应用于基因治疗、转基因动物研制和基因修饰等领域。
33.本发明的附加方面和优点将在下面的描述中部分给出,部分将从下面的描述中变得明显,或通过本发明的实践了解到。
附图说明
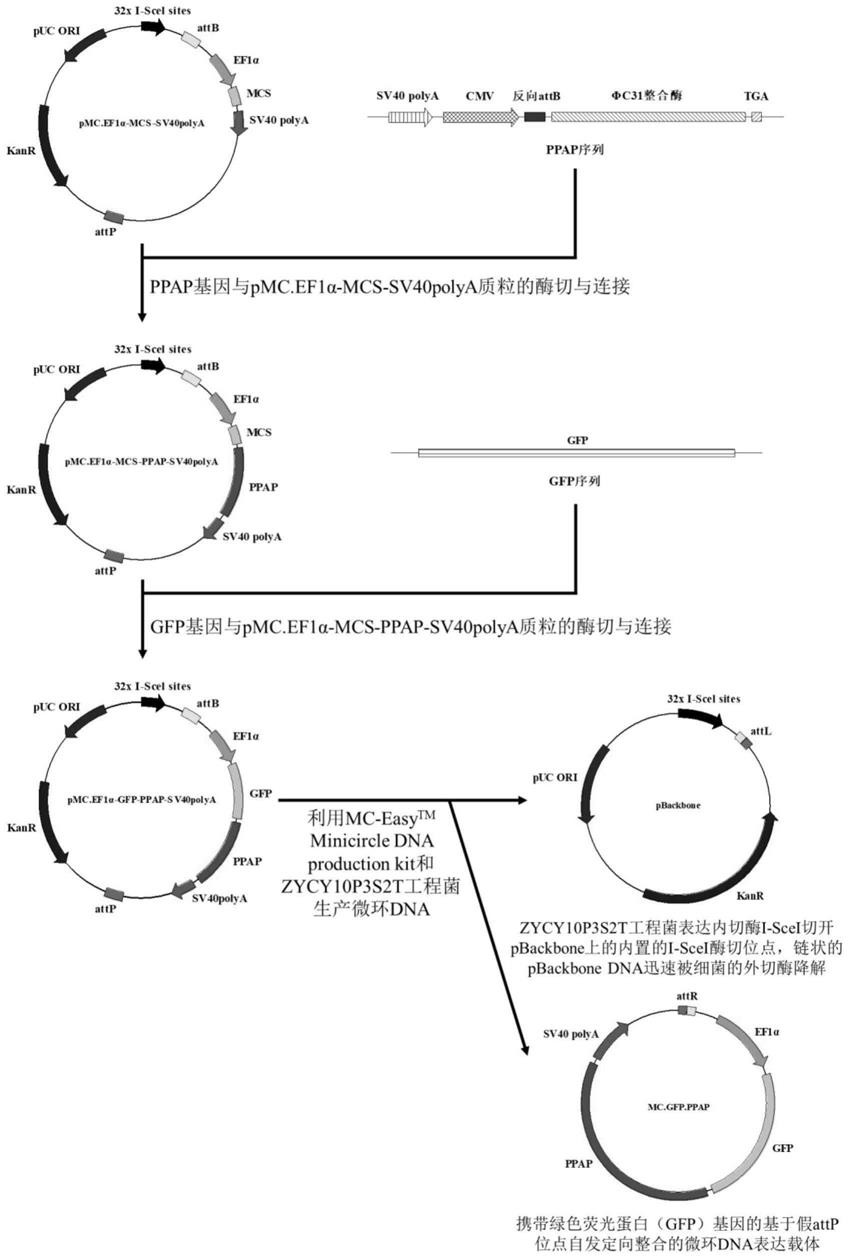
34.图1所示为基于假attp位点自发定向整合的通用型微环dna表达载体的构建过程示意图。
35.图2所示为ppap序列模式图。
36.图3所示为pmc.ef1α
‑
mcs
‑
ppap
‑
sv40polya质粒的质粒图谱。
37.图4所示为pmc.ef1α
‑
gfp
‑
ppap
‑
sv40polya质粒的质粒图谱。
38.图5所示为mc.gfp.ppap微环dna的图谱。
39.图6所示为微环dna转染hek293细胞48小时后,对照组和实验组细胞中绿色荧光蛋白的表达情况。
40.图7所示为微环dna转染hek293细胞10天后,对照组和实验组细胞中绿色荧光蛋白的表达情况。
41.图8所示为对照组和实验组hek293细胞基因组dna中绿色荧光蛋白基因的pcr产物琼脂糖凝胶电泳结果。
具体实施方式
42.下面结合实施例对本发明作进一步说明,但并不因此而限制本发明,需要说明的是,在不相冲突的前提下,以下描述的各实施例之间或各技术特征之间可以任意组合形成新的实施例。
43.本发明提供一种基于假attp位点自发定向整合的通用型微环dna表达载体,其重组母质粒是在微环dna空质粒的多克隆位点(mcs)中插入ppap序列组合而成,该重组母质粒在zycy10p3s2t工程菌内发生分子内位点特异性重组而生产出具有自发定向整合假attp位点功能的微环dna表达载体。
44.所述的微环dna空质粒优选pmc.cmv
‑
mcs
‑
sv40polya质粒或pmc.ef1α
‑
mcs
‑
sv40polya质粒,更优选pmc.ef1α
‑
mcs
‑
sv40polya质粒。
45.所述的ppap序列包括依次连接的polya加尾信号、启动子、反向attb序列、φc31整合酶基因和终止密码子。
46.所述的polya加尾信号优选牛生长激素多聚核苷酸bpa或sv40加尾信号,更优选sv40加尾信号,所述的sv40加尾信号核苷酸序列如seq.id.no.1所示。
47.本发明采用的polya加尾信号可以为bpa、sv40等加尾信号之一,sv40加尾信号只是本发明构建的ppap序列中的更优选polya加尾信号,本发明可以根据表达宿主细胞的不同、ppap序列插入的微环dna空质粒的不同等条件选择不同的polya加尾信号构建ppap序列。
48.所述的启动子优选巨细胞病毒cmv启动子、劳斯肉瘤病毒rsv启动子、泛素ubc启动子或延长因子ef1α启动子,更优选巨细胞病毒cmv启动子,所述巨细胞病毒cmv启动子核苷酸序列如seq.id.no.2所示。
49.本发明采用的启动子可以为cmv、rsv、ubc和ef1α等真核表达启动子之一,cmv启动
子只是本发明构建的ppap序列中的更优选启动子,本发明可以根据表达宿主细胞的不同、ppap序列插入的微环dna空质粒的不同等条件选择不同的启动子构建ppap序列。
50.所述的反向attb序列的核苷酸序列如seq.id.no.3所示。
51.所述的φc31整合酶基因核苷酸序列如seq.id.no.4所示。
52.所述的终止密码子核苷酸序列为tga。
53.所述的ppap序列的完整核苷酸序列如seq.id.no.5所示。
54.所述的zycy10p3s2t工程菌购自sbi公司。
55.一种基于假attp位点自发定向整合的通用型微环dna表达载体及其重组母质粒的构建方法,包括以下步骤:
56.(1)采用全基因序列合成的方式合成dna片段,该dna片段包括依次连接的bstbi酶切位点、ppap序列和bamhi酶切位点,所述的ppap序列的完整核苷酸序列如seq.id.no.5所示;
57.(2)采用bstbi和bamhi内切酶对步骤(1)所得的dna片段进行双酶切,并回收酶切后的含ppap序列的dna片段;
58.(3)采用bstbi和bamhi内切酶对pmc.ef1α
‑
mcs
‑
sv40polya质粒进行双酶切,并回收酶切后的线性质粒;
59.(4)采用dna连接酶连接步骤(2)双酶切后回收的dna片段和步骤(3)双酶切后回收的线性质粒,获得含ppap序列的微环dna重组母质粒pmc.ef1α
‑
mcs
‑
ppap
‑
sv40polya,该pmc.ef1α
‑
mcs
‑
ppap
‑
sv40polya质粒即为基于假attp位点自发定向整合的通用型微环dna表达载体的重组母质粒;
60.(5)将需要整合入宿主细胞染色体的外源基因片段插入到步骤(4)所得的微环dna重组母质粒pmc.ef1α
‑
mcs
‑
ppap
‑
sv40polya的多克隆位点(mcs)并进行扩增,其中,所述的外源基因片段可采用全基因序列合成或pcr克隆等方法获得,本发明可根据实际具体情况选择最优的方法获得需要整合入宿主细胞染色体的外源基因片段,所述的pmc.ef1α
‑
mcs
‑
ppap
‑
sv40polya质粒的多克隆位点(mcs)包括的限制性内切酶识别位点为:xbai、nhei和ecori;
61.(6)将步骤(5)所得的携带需要整合入宿主细胞染色体的外源基因片段的微环dna重组母质粒转化zycy10p3s2t工程菌,按照mc
‑
easy
tm minicircledna production kit操作说明书,扩增和诱导转化后的zycy10p3s2t工程菌,然后采用常规质粒提纯试剂盒提纯由zycy10p3s2t工程菌生产出的微环dna,该微环dna即为基于假attp位点自发定向整合的通用型微环dna表达载体,所述的微环dna为具有attr位点、延长因子ef1α启动子、需要整合入宿主细胞染色体的外源基因片段、ppap序列和sv40polya加尾信号的环状dna。
62.一种基于假attp位点自发定向整合的通用型微环dna表达载体的应用,包括将含有ppap序列的携带需要整合入宿主细胞染色体的外源基因片段的微环dna表达载体转染宿主细胞,其中所述的含有ppap序列的携带需要整合入宿主细胞染色体的外源基因片段的微环dna表达载体即为基于假attp位点自发定向整合的通用型微环dna表达载体。
63.所述的转染宿主细胞,转染方法优选化学转染方法或物理转染方法,本发明可以根据宿主细胞的不同选择不同的转染方法。
64.所述的宿主细胞优选真核细胞,更优选人细胞。
65.本发明所用的原料或试剂除特别说明之外,均市售可得。
66.在本发明的下述示例性的实施例中,所使用的质粒载体为pmc.ef1α
‑
mcs
‑
sv40polya质粒、所使用的工程菌为zycy10p3s2t工程菌(大肠杆菌e.coli)、mc
‑
easy
tm minicircle dna production kit,这些材料均购自sbi公司,所使用的菌株e.coli top10购自invitrogen公司,所使用的pcr缓冲液、dntp混合物、dna聚合酶均购自takara公司,其余所使用的试剂均为市售商品。在本发明提供的示例性实施例中,目的基因为绿色荧光蛋白(gfp)。
67.实施例1表达载体的构建
68.如图1所示,本实施例提供了一种基于假attp位点自发定向整合的通用型微环dna表达载体及其构建方法,包括以下步骤:
69.(1)采用全基因序列合成的方式合成依次连接的bstbi酶切位点、ppap序列(seq.id.no.5)和bamhi酶切位点核苷酸序列;
70.本实施例所述的ppap序列模式图如图2所示;
71.本实施例所述依次连接的各核苷酸序列仅为全基因合成的基因(即dna片段)双链中的一条,另一条链与依次连接的各核苷酸序列互补;
72.(2)用bstbi和bamhi对步骤(1)合成的核苷酸序列进行双酶切,采用琼脂糖凝胶电泳鉴定酶切结果,并回收酶切后的核苷酸片段;
73.(3)用bstbi和bamhi对pmc.ef1α
‑
mcs
‑
sv40polya质粒载体进行双酶切,采用琼脂糖凝胶电泳鉴定酶切结果,并回收线性pmc.ef1α
‑
mcs
‑
sv40polya质粒载体;
74.(4)采用t4dna连接酶将步骤(2)所得酶切后的核苷酸片段和步骤(3)所得的酶切后的线性pmc.ef1α
‑
mcs
‑
sv40polya质粒载体按3∶1的摩尔比在16℃下进行连接过夜或者室温下连接2小时,然后将连接产物加入e.coli top10菌株感受态细胞悬液中,进行转化,并接种在含有卡那霉素的lb平板上37℃培养过夜,挑取单菌落进行重组质粒载体的双酶切(bstbi和bamhi)验证;选取酶切结果验证的阳性克隆,进行测序验证,将插入核苷酸片段和步骤(1)合成的所述核苷酸序列完全一致的重组质粒载体命名为pmc.ef1α
‑
mcs
‑
ppap
‑
sv40polya,该pmc.ef1α
‑
mcs
‑
ppap
‑
sv40polya质粒即为基于假attp位点自发定向整合的通用型微环dna表达载体的重组母质粒,其质粒图谱如图3所示;
75.步骤(4)所得的基于假attp位点自发定向整合的通用型微环dna表达载体的重组母质粒其质粒图谱如图3所示,其中包括puc ori、kanr、32
×
i
‑
scei sites、attb序列、ef1α启动子、多克隆位点(mcs)、ppap序列、sv40 polya和attp序列;puc ori是质粒的复制原点,为了质粒能在细菌里复制扩增;kanr是卡那霉素抗性基因,用于筛选携带质粒的细菌;32
×
i
‑
scei sites是32个i
‑
scei酶切位点,是为了能将生产微环dna后剩余的重组母质粒和含有attl位点的细菌质粒dna环切开降解;ef1α启动子用于启动下游多克隆位点(mcs)中插入的外源基因的表达;多克隆位点(mcs)包含3个唯一的限制性内切酶识别位点,分别为xbai、nhei和ecori,用于外源基因片段的插入;ppap序列使微环dna具有自发定向整合假attp位点的功能;sv40 polya为ppap序列中的φc31整合酶基因提供加尾信号;attb序列和attp序列使重组母质粒在φc31整合酶的介导下形成含有attl位点的细菌质粒dna环和含有attr位点的微环dna;
76.(5)采用全基因序列合成或pcr克隆等方法获得需要整合入宿主细胞染色体的外
源基因片段,应当指出,本发明可根据实际具体情况选择最优的方法获得需要整合入宿主细胞染色体的外源基因片段,本实施例为简要说明本发明提供的基于假attp位点自发定向整合的通用型微环dna表达载体的应用,选择编码绿色荧光蛋白的基因序列作为需要整合入宿主细胞染色体的外源基因片段,并采用全基因序列合成的方式合成依次连接的xbai酶切位点、绿色荧光蛋白序列(seq.id.no.6)和ecori酶切位点核苷酸序列;
77.(6)用xbai和ecori对步骤(5)合成的核苷酸序列进行双酶切,采用琼脂糖凝胶电泳鉴定酶切结果,并回收酶切后的核苷酸片段;
78.(7)用xbai和ecori对步骤(4)所得的pmc.ef1α
‑
mcs
‑
ppap
‑
sv40polya重组质粒载体进行双酶切,采用琼脂糖凝胶电泳鉴定酶切结果,并回收线性pmc.ef1α
‑
mcs
‑
ppap
‑
sv40polya重组质粒载体;
79.(8)采用t4dna连接酶将步骤(6)所得酶切后的核苷酸片段和步骤(7)所得的酶切后的线性pmc.ef1α
‑
mcs
‑
ppap
‑
sv40polya重组质粒载体按3∶1的摩尔比在16℃下进行连接过夜或者室温下连接2小时,然后将连接产物加入e.coli top10菌株感受态细胞悬液中,进行转化,并接种在含有卡那霉素的lb平板上37℃培养过夜,挑取单菌落进行重组质粒载体的双酶切(xbai和ecori)验证;选取酶切结果验证的阳性克隆,进行测序验证,将插入核苷酸片段和步骤(5)合成的所述核苷酸序列完全一致的重组质粒载体命名为pmc.ef1α
‑
gfp
‑
ppap
‑
sv40polya,该pmc.ef1α
‑
gfp
‑
ppap
‑
sv40polya重组质粒载体的质粒图谱如图4所示;
80.(9)按照mc
‑
easy
tm minicircle dna production kit操作说明书,利用mc
‑
easy
tm minicircle dna production kit、zycy10p3s2t工程菌和步骤(8)所得的pmc.ef1α
‑
gfp
‑
ppap
‑
sv40polya重组质粒载体生产微环dna,步骤简述如下:
81.(9a)将步骤(8)所得的pmc.ef1α
‑
gfp
‑
ppap
‑
sv40polya重组质粒载体转化zycy10p3s2t工程菌,并接种在含有卡那霉素的lb平板上37℃培养过夜;
82.(9b)将步骤(9a)所得的单菌落接种于稀释后的生长培养基中,30℃摇晃(250次/分钟)培养过夜;
83.(9c)经过大约16小时的过夜培养后,向步骤(9b)所得的菌液中加入诱导培养基,30℃摇晃(250次/分钟)培养5
‑
5.5小时,在该过程中,zycy10p3s2t工程菌被诱导表达φc31整合酶和内切酶i
‑
scei,其中,φc31整合酶介导形成含有attl位点的细菌质粒(pbackbone)dna环和含有attr位点的微环dna;内切酶i
‑
scei切开pbackbone上的内置的i
‑
scei酶切位点,链状的pbackbonedna迅速被细菌的外切酶降解,微环dna成为唯一的染色体外环状dna;
84.(10)采用常规质粒提纯试剂盒提纯步骤(9c)所得的含attr位点、延长因子ef1α启动子、绿色荧光蛋白基因序列、ppap序列和sv40polya加尾信号的微环dna,即mc.gfp.ppap,该mc.gfp.ppap的质粒图谱如图5所示。
85.步骤(10)所得的mc.gfp.ppap微环dna含有ppap序列,该ppap序列中包括sv40加尾信号、cmv启动子、反向attb序列、φc31整合酶基因和终止密码子tga,其中,sv40加尾信号作为上游需要整合入宿主细胞染色体的外源基因片段(本实施例中为绿色荧光蛋白基因)的polya加尾信号;cmv启动子、反向attb序列、φc31整合酶基因、终止密码子tga和下游的sv40 polya加尾信号共同组成mc.gfp.ppap微环dna上的φc31整合酶基因表达盒,当mc.gfp.ppap微环dna转染进入宿主细胞后,该φc31整合酶基因表达盒便在宿主细胞内表
达φc31整合酶,该φc31整合酶基因表达盒中的反向attb序列在该φc31整合酶的介导下可与宿主细胞染色体基因组中“假attp位点”发生同源重组,从而使得mc.gfp.ppap微环dna位点特异性地整合入真核细胞染色体基因组中的转录活跃区,有利于外源基因(本实施例中为绿色荧光蛋白基因)持续高效表达,与此同时,该φc31整合酶基因表达盒中的cmv启动子和φc31整合酶基因从反向attb序列处分离并分别位于被“打开”的mc.gfp.ppap微环dna的两端,使得该φc31整合酶基因在mc.gfp.ppap微环dna整合入宿主细胞染色体后停止表达。
86.实施例2表达载体的应用
87.本实施例提供了一种基于假attp位点自发定向整合的通用型微环dna表达载体的应用,所使用的基于假attp位点自发定向整合的通用型微环dna表达载体为实施例1所得的mc.gfp.ppap微环dna,具体应用步骤如下:
88.(1)细胞铺板
89.将hek293细胞接种于6孔板中,待细胞生长至60%
‑
80%汇合率时进行转染。
90.(2)转染
91.(2a)在转染前2小时,移除hek293细胞上原有的培养基,换为新鲜的含有10%胎牛血清的dmem培养基;
92.(2b)将2μg微环dna用100μl无血清dmem培养基稀释,充分混匀后制成dna稀释液,其中,对照组用只含绿色荧光蛋白基因的mc.gfp微环dna,实验组用实施例1所得的mc.gfp.ppap微环dna;
93.步骤(2b)中所述的只含绿色荧光蛋白基因的mc.gfp微环dna是在pmc.ef1α
‑
mcs
‑
sv40polya质粒载体上的多克隆位点(mcs)中只插入绿色荧光蛋白基因片段,而后利用zycy10p3s2t工程菌和mc
‑
easy
tm minicircle dna production kit生产所得;
94.(2c)向dna稀释液中直接加入2μl neofect
tm
转染试剂,轻柔混匀,室温静置15
‑
30分钟,转染复合物制备完成;
95.(2d)将转染复合物加入hek293细胞培养基中,轻柔混匀。
96.(3)培养细胞48小时后,将对照组和实验组细胞分别置于倒置荧光显微镜下观察,结果如图6所示:由图6所示结果可知,对照组和实验组的hek293细胞在蓝色激发光下均观察到绿色荧光,这表明对照组的mc.gfp微环dna和实验组的mc.gfp.ppap微环dna均转染成功并在细胞中表达绿色荧光蛋白;观察结束后,将对照组和实验组的hek293细胞放回孵箱,继续培养。
97.(4)培养细胞10天后,将对照组和实验组细胞分别置于倒置荧光显微镜下观察,结果如图7所示;由图7所示结果可知,对照组hek293细胞在蓝色激发光下无绿色荧光,实验组hek293细胞在蓝色激发光下仍能观察到绿色荧光,这说明转染只含绿色荧光蛋白基因的mc.gfp微环dna的对照组hek293细胞已无绿色荧光蛋白表达,而转染mc.gfp.ppap微环dna的实验组hek293细胞持续表达绿色荧光蛋白。
98.(5)为进一步验证实验组hek293细胞中的mc.gfp.ppap微环dna已自发定向整合入细胞染色体基因组中,故采取以下方法:
99.(5a)采用常规哺乳动物基因组dna抽提试剂盒抽提步骤(4)中所述的对照组和实验组hek293细胞的基因组dna;
100.(5b)pcr
101.利用vector nti设计绿色荧光蛋白基因的引物,如下:
102.正向引物:5
’‑
aactgttcactggcgtggt
‑3’
103.反向引物:5
’‑
tcacaaactccagcagga
‑3’
104.按如下pcr反应体系与反应条件进行pcr反应:
105.pcr反应体系:pcr缓冲液(2
×
primestar gc buffer)25μl,dntp混合物4μl,正向引物反向引物各1μl(引物浓度10μm),模板(抽提的hek293细胞基因组dna)1μl,dna聚合酶(primestar hs dna polymerase)0.5μl,双蒸水17.5μl
106.pcr反应条件:98℃预变性10秒;(98℃5秒,68℃102秒)
×
30个循环;72℃5分钟
107.(5c)将步骤(5b)所得的pcr反应产物进行琼脂糖凝胶电泳,结果如图8所示,其中,泳道1为对照组hek293细胞的基因组dna的pcr反应产物,在该泳道未见条带,说明对照组hek293细胞的基因组dna中无绿色荧光蛋白基因;泳道2为实验组hek293细胞的基因组dna的pcr反应产物,在该泳道可见长度约为700bp的单一条带,说明实验组hek293细胞的基因组dna中含有绿色荧光蛋白基因,这表明转染入实验组hek293细胞中的mc.gfp.ppap微环dna已自发定向整合入宿主细胞染色体基因组中。
108.尽管已经示出和描述了本发明的实施例,本领域的普通技术人员可以理解:在不脱离本发明的原理和宗旨的情况下可以对这些实施例进行多种变化、修改、替换和变型,本发明的范围由权利要求及其等同物限定。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1