一种多环芳基化合物在抗真菌药物制备中的应用
1.本发明属于药物领域。更具体而言,本发明涉及一种多环芳基化合物s1在制备用于抗念珠菌、曲霉、毛霉、隐球菌为代表的致病真菌药物中的应用。
背景技术:
2.真菌感染严重威胁着人类的健康生活,真菌感染根据侵犯人体的部位可分为浅部真菌病与深部真菌病,后者主要病原体为念珠菌、曲霉、毛霉、隐球菌为代表的条件致病真菌,每年可造成超过1300万人感染、160万人死亡,其死亡人数已超过了疟疾、结核以及乳腺癌致死数量。更为严峻的是,即使接受规范抗真菌治疗,在免疫抑制患者中,其死亡率也高达67%。
3.目前,深部真菌感染以药物治疗为主,但可用药物十分有限,主要是唑类(氟康唑、伏立康唑等三唑类)、多烯类(两性霉素b)、棘白菌素类(卡泊芬净、米卡芬净)三类。这些药物各有优缺点,唑类虽然安全性与口服生物利用率高,但其抑菌作用导致耐药严重;多烯类抗菌谱广、疗效确切但具有显着的肝、肾毒性,约30%接受两性霉素b治疗的成年人发生急性肾损伤,导致死亡率,住院时间和费用均显着增加;棘白菌素类是30年来批准的新一类抗真菌药物,但口服的生物利用率低。因此,治疗深部真菌感染的药物选择局限,无法满足日益增长的临床需求,迫切需要开发新型抗真菌药物。
技术实现要素:
4.本发明为了克服现有抗真菌药物的不足,提供一种用于制备抗真菌感染的多环芳基化合物s1,所述的多环化合物结构式为:
[0005][0006]
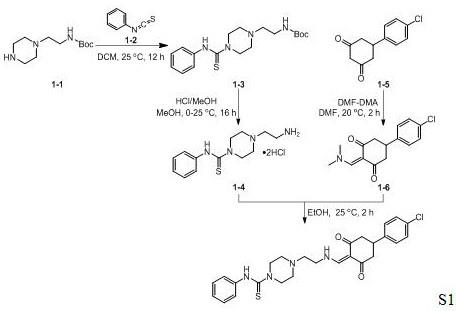
本发明涉及一种制备多环芳基化合物s1的方法:化合物1-1与化合物1-2反应生成硫脲化合物1-3。后者在盐酸的条件下脱掉boc保护基生成盐酸盐化合物1-4。化合物1-5与n,n-二甲基甲酰胺二甲基缩醛反应生成二酮化合物1-6。在乙醇为溶剂的条件下,化合物1-4与化合物1-6反应得到多环化合物s1。
[0007][0008]
本发明还涉及一种药物组合物,其含有治疗有效剂量的s1所示的多环芳基化合物其药学上可接受的盐,以及一种或多种药学上可接受的载体所组成的药物组合物。
[0009]
本技术所述的“可药用盐”是指本发明化合物的盐,这类盐用于哺乳动物体内时具有安全性和有效性,且具有应有的生物活性,通常以游离酸的形式使用。可用本领域公知的方法制备本发明的游离氨基化合物的酸加合盐,并可从有机酸和无机酸制备。适合的有机酸包括马来酸、反丁烯二酸、安息香酸、抗坏血酸、琥珀酸、甲磺酸、乙酸、三氟乙酸、草酸、丙酸、酒石酸、水杨酸、柠檬酸、葡萄糖酸、乳酸、扁桃酸、苯乙酸、天冬氨酸、硬脂酸、棕榈酸、乙二醇酸、谷氨酸、甲苯磺酸和苯磺酸。适合的无机酸包括盐酸、氢溴酸、硫酸、磷酸和硝酸。因此,本发明化合物的“药物可接受的盐”应包括和所有可接受的盐形式。
[0010]
本发明还涉及含有治疗有效剂量的s1所示的多环芳基化合物,或其药学上可接受的盐,或其药物组合物在制备用于抗念珠菌、曲霉、隐球菌等致病真菌感染药物中的用途。
[0011]
本发明还涉及含有治疗有效剂量的s1所示的多环芳基化合物,或其药学上可接受的盐,或其所述的药物组合物在制备用于治疗真菌感染所引起的浅部及深部真菌感染疾病药物中的用途。
[0012]
本技术所述的“药物组合物”表示含有一种或多种本文所述化合物或其生理学上/可药用的盐或前体药物与其他化学组分的混合物,以及其他组分例如生理学/可药用的载体和赋形剂。药物组合物的目的是促进对生物体的给药,利于活性成分的吸收进而发挥生物活性。
[0013]
本技术具有的有益效果:
[0014]
1、本技术所述化合物s1对包括但不限于念珠菌、曲霉、毛霉等的致病真菌具有广谱抗真菌作用;
[0015]
2、本技术所述化合物s1对唑类耐药的念珠菌有效;
[0016]
3、本技术所述化合物s1对哺乳动物毒性低;
[0017]
4、本技术所述化合物s1可通过口服、注射的方式治疗系统性真菌感染;
[0018]
5、本技术所述化合物s1可通过局部应用的方式治疗黏膜真菌感染。
附图说明
[0019]
图1为化合物s1的1h-nmr核磁图谱图;
[0020]
图2为化合物s1治疗蜡螟真菌感染的生存率变化图;
[0021]
图3为化合物s1治疗阴道念珠菌感染的载菌量变化图;
[0022]
图4为化合物s1治疗系统性真菌感染小鼠的生存率变化图;
[0023]
图5为化合物s1治疗系统性真菌感染小鼠的器官载菌量变化图;
[0024]
图6为化合物s1治疗系统性真菌感染小鼠的肾脏病理图。
具体实施方式
[0025]
本技术所述的s1化合物的结构是通过核磁共振(nmr)或/和质谱(ms)来确定的。
[0026]
nmr位移(δ)以10-6
(ppm)的单位给出,nmr的测定是用bruker avance-400核磁仪,测定溶剂为氘代氯仿(cdcl3)、内标为四甲基硅烷(tms)。
[0027]
柱层析一般使用烟台黄海硅胶200~300目硅胶为载体。
[0028]
本发明的已知的起始原料可以采用或按照本领域已知的方法来合成,或可购买自aldrich chemical company,韶远化学科技(accela chembio inc),百灵威,安耐吉,达瑞化学品等公司。
[0029]
实施例中无特殊说明,反应能够均在氩气氛或氮气氛下进行。
[0030]
氩气氛或氮气氛是指反应瓶连接一个约1l容积的氩气或氮气气球。
[0031]
实施例中无特殊说明,溶液是指水溶液。
[0032]
实施例中无特殊说明,反应的温度为室温,为20℃~30℃。
[0033]
实施例1化合物1-3的合成
[0034][0035]
常温下,将化合物1-1(5.00g,21.8mmol,1.00eq)溶于二氯甲烷(25ml)中,随后加入化合物1-2(3.54g,26.2mmol,3.13ml,1.20eq)。搅拌反应12小时后,将反应液过滤后进行浓缩得粗产物。所得粗产物通过硅胶柱层析(dcm:meoh=8:1)纯化得到白色国体化合物1-3。
[0036]
化合物1-3:1h-nmr(400mhz,cdcl3),δ7.42(s,1h),7.34-7.30(t,j=7.8hz,2h),7.16(s,1h),7.14-7.10(t,j=7.6hz,2h),4.91(s,1h)3.83-3.80(t,j=4.8hz,4h),3.22-3.21(d,j=5.2hz,2h),2.50-2.46(m,6h),1.45(s,9h);lc-ms m/z。
[0037]
实施例2化合物1-4的合成
[0038][0039]
将化合物1-3(12.0g,32.9mmol,1.00eq)溶于甲醇(60.0ml)中,在零度条件下,加入盐酸甲醇溶液(32.9mmol,48.0ml,1.00eq)。随后,将反应升至常温进行反应。反应16小时
后,反应液在减压条件下浓缩得到白色固体粗盐酸盐化合物1-4(13.1g)。
[0040]
化合物1-4:lc-ms m/z 265.2[m+h]
+
。
[0041]
实施例3化合物1-6的合成
[0042][0043]
常温下,将化合物1-5(8.00g,35.9mmol,1.00eq)溶于n,n-二甲基甲酰胺(100ml)中,随后加入n,n-二甲基甲酰胺二甲基缩醛(35.9g,301mmol,40.0ml,8.38eq)。搅拌反应2小时后,将反应液进行过滤得到滤液。滤液在减压条件下旋干浓缩得到到淡黄色固体化合物1-6(8.50g,30.6mmol,产率为85.2%)。
[0044]
化合物1-6:1h nmr(400mhz,cdcl3),δ8.08(s,1h),7.32-7.30(m,2h),7.19-7.17(d,j=8.4hz,2h),3.43(s,3h),3.37-3.32(m,1h),3.22(s,3h),2.77-2.62(m,4h);lc-ms m/z 278.1[m+h]
+
。
[0045]
实施例4化合物s1的合成
[0046][0047]
常温下,将盐酸盐化合物1-4(13.1g,38.8mmol,1.20eq.)溶于乙醇(52.0ml)中,随后加入化合物1-6(8.99g,32.4mmol,1.00eq)进行反应。搅拌反应2小时后,加入氯仿萃取三次(100ml x 3),并用饱和食盐水洗涤(100ml x 1)。将有机相合并过滤后,在减压条件下浓缩得到粗产物。所得粗产物经硅胶柱层析纯化得到白色固体化合物s1(11.2g,22.5mmol,产率为69.6%)。化合物s1的1h-nmr核磁图谱见图1。
[0048]
化合物s1:1h nmr(400mhz,cdcl3),δ11.24-11.21(brs,1h),8.20-8.16(d,j=14.4hz,1h),7.36-7.30(m,5h),7.18-7.16(m,3h),7.11-7.10(d,j=7.6hz,2h),3.88-3.86(t,j=4.4hz,4h),3.54-3.51(m,2h),3.36(m,1h),2.73-2.72(m,3h),2.66-2.64(m,3h),2.59-2.56(t,j=4.8hz,4h),2.05-2.02(t,j=6.4hz,1h);lc-ms m/z 497.2[m+h]
+
。
[0049]
实施例5化合物s1体外抗真菌活性评价
[0050]
以下结合实施例进一步描述解释本发明,但这些实施例并非意味着限制本发明的范围。
[0051]
(1)菌株与培养基
[0052]
白念珠菌标准株(sc5314)、白念珠菌临床株(ca1-3)、近平滑念珠菌标准株(atcc 22019)、克柔念珠菌标准株(atcc2 8870,)、热带念珠菌标准株(atcc 750,);烟曲霉标准株(atcc 1022)、烟曲霉临床株(af1-3);毛霉(nrrl3631);新生隐球菌标准株(atcc 32609);肺孢子菌标准株(atcc pra-159)。菌株置于ypd+甘油培养基中-80℃保存,ypd琼脂平板上传代培养,ypd液体培养基中活化。
[0053]
(2)实验方法
[0054]
采用微量液基稀释法检测化合物s1对上述真菌的最低抑菌浓度(mic
50
)。菌株用yepd琼脂培养基35℃培养24h,活化2次后用接种环挑取活化的白念珠菌,rpmi 1640培养液稀释,利用细胞计数板计数,将菌悬液的浓度调整为所需工作浓度(2.0~2.5)
×
103cfu/ml,使用前新鲜配制。用rpmi 1640液体培养基将药物储备液稀释至2倍工作浓度。取其100μl倍比稀释后的药液一次加入96孔板的第2-11列;第1列为生长对照孔(不加药物,以100μl的rpmi 1640培养基代替),第12列为空白对照孔(只加入200μl的rpmi 1640培养基)。96孔板的第1-11列加入100μl的菌悬液至工作浓度。加入菌液和药物的平板在35℃培养48h后应用酶标仪检测波长490nm的od值。每个孔均减去背景读数,与无药菌悬液孔结果对比,以抑制50%真菌生长的最低药物浓度,作为最终mic
50
结果。实验重复3次。
[0055]
(3)实验结果
[0056]
本发明中化合物的生物活性通过以上的实验方法进行测定,测得的化合物体外抗真菌活性结果如表1所示,化合物s1具有广谱抗真菌活性。
[0057]
表1.化合物s1体外抗真菌mic
50
(μg/ml)
[0058][0059]
化合物s1细胞毒性评价
[0060]
实施例6
[0061]
(1)实验方法
[0062]
lo-2与raw264.7细胞在dmem+10%fbs+1%青/链霉素的培养基中培养;hk-2在dmem/f-12+10%fbs+1%青/链霉素的培养基中培养;
[0063]
cck-8法检测生长毒性:将5
×
104cfu/ml的细胞100μl加入96孔板中,放置在37℃,5%co2的培养箱中预培养24h。向每个培养孔里依此加入不同浓度梯度的小分子化合物,将96孔板放入37℃,5%co2的培养箱中培养6h。6h后向待测样品中加入cck-8溶液10μl。将96孔板放在培养箱内孵育1-4h。用多功能酶标仪测定在450nm处的吸光度。结果取3次独立实验的平均数来表示。
[0064]
ldh法检测破坏毒性:将5
×
104cfu/ml的细胞100μl加入96孔板中,放置在37℃,5%co2的培养箱中过夜。更换培养基100μl。向每个培养孔里依此加入不同浓度梯度的小分子化合物培养24h。在阳性对照孔内加入10μl lysis buffer后,在37℃,5%co2的培养箱中培养30min。在每孔中加入100μl working solution后,在避光、室温的条件下培养30min。在每孔中加入50μl stop solution后立刻用多功能酶标仪检测490nm的吸光度。按照公式计算ldh释放百分比。结果取3次独立实验的平均数来表示。
[0065]
(2)实验结果
[0066]
结果如下表2所示,24h小分子s1对lo-2、hk-2、raw264.7生长的ic
50
值分别为78.44、73.61、86.57μg/ml,48h小分子对lo-2、hk-2、raw264.7生长的ic
50
值分别为53.91、105.21、35.64μg/ml,显著高于s1对致病真菌的mic
50 10倍以上。
[0067]
对细胞的破坏毒性显示,s1造成50%细胞破坏需要比抑制生长更大的剂量,在lo-2、hk-2、raw264.7细胞中分别需要131.91、106.1、68.74μg/ml。上述结果说明该小分子在念珠菌和哺乳动物之间具有选择性。
[0068]
表2 s1对肝、肾、巨噬细胞的ic
50
结果(μg/ml)
[0069][0070]
化合物s1在体内抗真菌活性评价
[0071]
以白念珠菌为例,对化合物s1进行体内抗真菌活性评价。
[0072]
实施例7化合物s1蜡螟体内抗真菌活性评价
[0073]
(1)实验方法
[0074]
首先选取最后一龄的蜡螟幼虫(长度约3cm、体重约0.5g),尽量选取体态健康,没有黑色杂点的幼虫。随机分组,每组20只,置于直径10cm的平皿中。将白念珠菌sc5314至于ypd液体培养基中活化至对数生长末期,收集菌体,pbs清洗两次。用生理盐水调整菌液浓度至1
×
108cfu/ml。取大蜡螟固定蜡螟,腹部朝上,将蜡螟幼虫进针部位酒精消毒,手持虫体,用汉密尔顿微量注射针在右下第二足轻轻刺穿,针头至虫体中间部位注射10μl菌液。造模成功后置于干净纸巾静置1min。左下第二足注射药物s1(25、50、100mg/kg),对照组wt注射同体积生理盐水。每天定时观察并移除死亡的虫体,记录各组蜡螟存活时间,3天后统计蜡螟生存率。
[0075]
(2)实验结果
[0076]
实验结果如图2所示。对照组(wt)蜡螟感染白念珠菌后第四天全部死亡,化合物s1
(25mg/kg)对蜡螟生存率无明显影响,化合物s1(50、100mg/kg)治疗后蜡螟生存率分别提高至18.3%与45%(p《0.05)。
[0077]
实施例8化合物s1局部治疗阴道念珠菌感染的药效
[0078]
(1)实验方法
[0079]
18-20g雌性balb/c小鼠在感染前三天腹腔注射100μl雌激素(100μl含0.5mgβ-雌二醇的芝麻油)以及100μl环磷酰胺(100mg/kg),之后每三天一次。感染当天,收集液体ypd中活化过夜的白念珠菌sc5314,pbs洗两次,调菌至菌液浓度为1
×
107cfu/ml。用水合氯醛将小鼠麻醉,每只小鼠阴道腔内注入10μl菌液,垫高小鼠臀部,静置半小时。感染后第二天,对应组别(每组3只)给与化合物s1 5μl(0.1、0.5、2.5mg),连续3填。第四天,用pbs灌洗小鼠阴道,稀释后抹于ypd平板上培养48h后计数。
[0080]
(2)实验结果
[0081]
结果如图3所示,局部治疗三天后,s1(2.5mg)组小鼠阴道载菌量下降两个梯度(p《0.05),s1(0.25、0.5mg)组小鼠阴道载菌量下降一个梯度。
[0082]
实施例9化合物s1口服与注射治疗系统性真菌感染小鼠的药效
[0083]
(1)实验方法
[0084]
将白念珠菌sc5314至于ypd液体培养基中活化至对数生长末期,收集菌体,pbs清洗两次。用生理盐水调整菌液浓度至2
×
106cfu/ml。将18-22g雌性balb/c小鼠随机分组,每组10只,分别是ns(注射生理盐水)、wt(注射sc5314)、wt+不同浓度药物治疗组(口服、注射)、单独药物对照组。分别将菌液经尾静脉注射至不同小鼠体内,每只100μl(造模菌量为2
×
105cells/只),ns组注射等体积的生理盐水。
[0085]
生存率:每天定时观察并记录各组小鼠生存状态,观察30天后计算生存率并将仍存活的小鼠处死。绘制生存曲线并通过log-rank test进行统计检验。
[0086]
病原学:于治疗结束后第1天,取每组小鼠(3只/组)左肾、脑、肝、脾组织,称重后研磨,将组织研磨液倍比稀释涂抹于ypd平皿上,30℃培养48h后统计菌落数量。结果采用student t test进行统计分析。
[0087]
组织病理学:将上述检测病原学指标的右肾置于10%甲醛固定,将其制作成he染色和pas染色的病理切片。
[0088]
(2)实验结果
[0089]
生存率变化结果如图4所示,化合物s1(100mg/kg)口服与注射均可将系统性感染小鼠生存率提高60%(p《0.05)。s1(50mg/kg)注射可将生存率提高10%,s1(50mg/kg)口服不影响生存率,但可延长系统性感染小鼠生存时间。
[0090]
器官载菌量结果如图5所示,化合物s1治疗7天结束后,小鼠肝、肾、脑中载菌量均下降2个梯度(p《0.05)。
[0091]
组织病理学结果如图6所示,小鼠肾、肝、脾、脑病理结果与ns对照无差异,未见组织损伤,未见菌丝、假菌丝、孢子和炎症细胞浸润。
[0092]
最后有必要说明的是,以上对本发明的具体实施例进行了详细描述,但其只作为范例,本发明并不限制于以上描述的具体实施例。对于本领域技术人员而言,任何对本发明进行的等同修改和替代也都在本发明的范畴之中。因此,在不脱离本发明的精神和范围下所作的均等变换和修改,都应涵盖在本发明的范围内。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1