一种通过血红蛋白催化交联的仿贻贝类水凝胶的制备方法
1.本发明属于生物催化领域,尤其涉及一种通过血红蛋白催化交联的仿贻贝类水凝胶的制备方法。
背景技术:
2.水凝胶是一种具有优越生物相容性的材料,它具有与人体体内细胞外基质类似的三维交联结构,因其具有一定的强度,良好的生物黏附及生物相容性等优异的理化性质和生物学性质。目前临床上较为广泛使用的粘合材料主要有合成材料氰基丙烯酸酯胶黏剂、聚氨酯,蛋白质仿生粘合材料纤维蛋白胶、明胶等。然而,这些材料在生物相容性、毒性或者粘附强度等方面并不十分理想。近年来,受贻贝启发的多酚类水凝胶得到了越来越多的关注。仿贻贝多酚类水凝胶含有大量的邻苯二酚或邻苯三酚基团,具有优良的粘附性、止血性和抑菌性,不仅可以实现伤口处的原位自组装和可控降解、适应伤口的尺寸与形状、提供伤口愈合所需的润湿环境,也可作为防护性的物理屏障,阻止外源微生物的入侵,促进创面组织愈合。
3.在仿贻贝类水凝胶的形成过程中,需要交联使聚合物形成三维多孔的网状结构。在以往的研究中通常使用化学氧化剂或辣根过氧化物酶(hrp)促进仿贻贝类水凝胶氧化交联。然而采用高碘酸钠(naio4)、氯化铁(fecl3)等化学氧化剂都具有一定的毒性,这些制备方法对凝胶的生物相容性或强度一定程度上会产生不利影响。使用hrp进行氧化交联虽然不产生有毒的副产物,所得到的凝胶具有良好的生物相容性,但由于其价格昂贵仍不适用于水凝胶的大规模生成应用。血红蛋白(hb)是脊椎动物红细胞内的呼吸蛋白,是自然界中一种来源十分广泛的蛋白质。由于其铁卟啉(fe
‑
heme)活性中心,而且具有天然的四级立体结构,可以用于酶催化反应,还显示出了比其它模拟酶(更高的催化活性。
4.为避免上述技术问题,本发明提供了一种通过血红蛋白催化交联的仿贻贝类水凝胶的制备方法以克服现有技术中的所述缺陷。
技术实现要素:
5.本发明的目的在于提供一种通过血红蛋白催化交联的仿贻贝类水凝胶的制备方法。
6.本发明是这样实现的,一种通过血红蛋白催化交联的仿贻贝类水凝胶的制备方法,步骤如下:
7.(1)称量60~200mg制备好的水凝胶前体放置于烧杯中,加入600~700μl磷酸盐缓冲盐溶液,备用;
8.(2)称取一定量的血红蛋白溶解于100μl磷酸盐缓冲盐溶液溶液中,备用;
9.(3)制备50μl浓度为25mmol/l含h
202
的磷酸盐缓冲盐溶液溶液,备用;
10.(4)用移液枪量取100μl步骤(2)中含血红蛋白的磷酸盐缓冲盐溶液溶液加入到步骤(1)中的溶液中,搅拌使之完全混合;
11.(5)用移液枪量取50μl步骤(3)中制备的含h
202
的磷酸盐缓冲盐溶液溶液加入到步骤(4)中搅拌后的混合溶液中;
12.(6)持续搅拌或静置步骤(5)中的溶液直至形成凝胶。
13.进一步的技术方案,所述步骤(1)中的磷酸盐缓冲盐溶液溶液的ph为7~8。
14.进一步的技术方案,步骤(1)中加入磷酸盐缓冲盐溶液到放有水凝胶前体的烧杯后,使用超声波细胞破碎仪对烧杯中的溶液超声处理一段时间,直至水凝胶前体充分溶解于磷酸盐缓冲盐溶液溶液中。
15.进一步的技术方案,所述步骤(1)中使用超声波细胞破碎仪超声处理溶液的时间为10~15分钟。
16.进一步的技术方案,步骤(2)中血红蛋白与磷酸盐缓冲盐溶液溶液的质量百分比为5%。
17.进一步的技术方案,所述步骤(6)中溶液放置的温度环境为30~50℃。
18.进一步的技术方案,所述步骤(6)中通过翻转试管法测试凝胶是否充分凝结并测定凝胶形成时间。
19.相较于现有技术,本发明的有益效果如下:
20.本发明提供的一种通过血红蛋白催化交联的仿贻贝类水凝胶的制备方法,本发明以血红蛋白作为制备水凝胶的交联剂,反应过程中不必使用有毒催化剂及其他有害成分,并且所得到的凝胶具有优异的生物相容性。该催化方法对环境友好,条件温和,成本低且凝胶效果也优于一般的酶催化或化学催化。
21.本发明制备的仿贻贝类水凝胶,具有良好的抗菌和止血效果。良好的生物相容性和可降解性,使其能够以粘附的方式应用于解决临床组织止血、组织愈合等问题。
附图说明
22.图1为壳聚糖
‑
咖啡酸衍生物(chi
‑
c)的反应式;
23.图2为水凝胶前体p(meo2ma
‑
oegma
‑
dma)的反应合成流程;
24.图3为本发明中实例1所得的水凝胶红外数据图;
25.图4为本发明中实例2所得的水凝胶红外数据图。
具体实施方式
26.为了使本发明的目的、技术方案及优点更加清楚明白,以下结合附图及实施例,对本发明进行进一步详细说明。应当理解,此处所描述的具体实施例仅仅用以解释本发明,并不用于限定本发明。
27.以下结合具体实施例对本发明的具体实现进行详细描述。
28.实施例一:
29.1.合成chi
‑
c水凝胶前体
30.水凝胶前体(壳聚糖
‑
咖啡酸衍生物)的制备过程:3g壳聚糖溶于100mlph值为5的盐酸溶液;2.37g咖啡酸和2.02gedc分别溶于25ml去离子水中;将咖啡酸和1
‑
(3
‑
二甲氨基丙基)
‑3‑
乙基碳二亚胺盐酸盐(edc)溶液缓慢加入壳聚糖溶液中;为了防止儿茶酚被氧化,溶液ph值保持在5.0。在混合液中加入50ml乙醇作为共溶剂室温下搅拌,以避免edc反应中
可能的沉淀;12小时后,在ph4.0的氯化钠溶液中透析2天,再在去离子水中透析4h,纯化得到产物壳聚糖
‑
咖啡酸衍生物(chi
‑
c);最后将产物冷冻干燥,使用前保存在无水分的干燥器中。反应式如图1。
31.2.制备chi
‑
c水凝胶
32.将60mg制备好的chi
‑
c溶于600μl且ph=7的磷酸盐缓冲盐溶液中,超声分散12分钟使水凝胶前体充分溶解;向水凝胶前体溶液中加入配置好的100μl质量分数为5wt%的血红蛋白的磷酸盐缓冲盐溶液,充分搅拌,混合均匀;在混合后的溶液中加入50μl浓度为25mmol/l含h2o2的磷酸盐缓冲盐溶液,40℃温度条件下搅拌直至混合液形成凝胶;用翻转试管法证实凝胶的形成。
33.实施例2
34.1.制备p(meo2ma
‑
oegma
‑
dma)水凝胶前体
35.将8g碳酸氢钠(nahco3)和20g四硼酸钠十水合物(na2b4o7·
10h2o)溶于200ml的二次蒸馏水中,并用针头插入溶液,氮气鼓泡30分钟以除去溶液中的氧气;随后,在溶液中加入10g盐酸多巴胺;将9.4ml甲基丙烯酸酐溶于50ml四氢呋喃中并用恒压漏斗逐滴加入到混合液中,用1m的氢氧化钠调节混合液的ph﹥8;室温搅拌反应16小时,反应后的混合液减压抽滤除去nahco3和na2b4o7·
10h2o固体;澄清透明的滤液用100ml乙酸乙酯洗涤,并用浓盐酸调节ph﹤2;随后将该溶液用100ml的乙酸乙酯萃取3次;将萃取后的300ml有机溶液加入无水硫酸镁(mgso4)密封于冰箱中冷藏干燥过夜;干燥后的溶液抽滤除去mgso4,旋蒸浓缩至50ml左右,在剧烈搅拌下逐滴滴入450ml冰正己烷中并将其冰箱中冷藏过夜重结晶,抽滤得到最终产物多巴胺甲基丙烯酰胺(dma)并于真空干燥箱中室温烘干。
36.通过自由基聚合合成水凝胶前体p(meo2ma
‑
oegma
‑
dma):3.00ml 2
‑
(2
‑
甲氧基乙氧基)甲基丙烯酸乙酯(meo2ma)、2.51ml寡聚(乙二醇)甲基醚甲基丙烯酸酯(oegma)、0.4803g dma和0.0625g偶氮二异丁腈(aibn)加入到装有15ml n,n
‑
二甲基甲酰胺(dmf)的圆底烧瓶中搅拌使之充分溶解;为了保护邻苯二酚结构反应中不被氧化,氮气鼓泡30分钟以保证除尽溶液中的氧气;于65℃的油浴中持续反应5h;反应结束后,反应液用四氢呋喃稀释,冰乙醚沉淀两次,室温真空干燥48h。反应流程如图2。
37.2.制备p(meo2ma
‑
oegma
‑
dma)水凝胶
38.将200mg制备好的水凝胶前体聚合物充分溶解在700μl且ph值为7的磷酸盐缓冲盐溶液中,超声分散12分钟使水凝胶前体充分溶解;向水凝胶前体溶液中加入100μl配置好的质量分数为5wt%的血红蛋白的磷酸盐缓冲盐溶液;充分搅拌,混合均匀;在混合后的溶液中加入50μl浓度为25mmol/l含h2o2的磷酸盐缓冲盐溶液,40℃温度条件下搅拌直至混合液形成凝胶;用翻转试管法证实凝胶的形成。
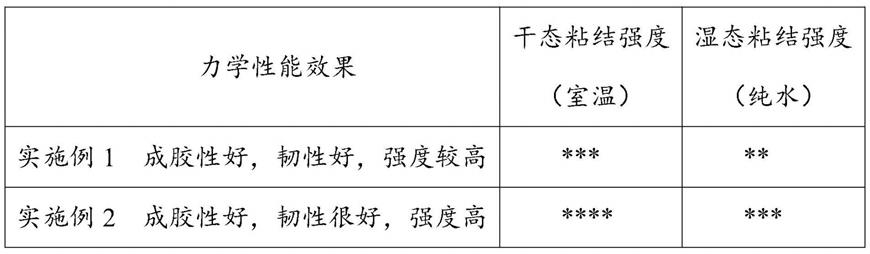
39.图3是实例1所得的水凝胶红外数据图,用来表征咖啡酸的儿茶酚基团成功接枝到壳聚糖上。图4是实例2所得的水凝胶红外数据图,用来表征meo2ma、oegma、dma三种单体的成功聚合。对实施例1和实施例2的水凝胶开展粘结砝码的实验:*代表能粘接小于50g砝码,**代表能粘接起100g砝码,***代表能粘接150g砝码,****代表能粘接起200g砝码,三种实施例中的水凝胶对人皮肤组织、玻璃、塑料、硬纸、橡胶、金属均有较好的粘结性。结果见表1(两种水凝胶的力学性能)。
[0040][0041]
表1
[0042]
以上所述仅为本发明的较佳实施例而已,并不用以限制本发明,凡在本发明的精神和原则之内所作的任何修改、等同替换和改进等,均应包含在本发明的保护范围之内。
[0043]
此外,应当理解,虽然本说明书按照实施方式加以描述,但并非每个实施方式仅包含一个独立的技术方案,说明书的这种叙述方式仅仅是为清楚起见,本领域技术人员应当将说明书作为一个整体,各实施例中的技术方案也可以经适当组合,形成本领域技术人员可以理解的其他实施方式。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1