2-氯-5-氯甲基噻唑连续化生产工艺的制作方法
2
‑
氯
‑5‑
氯甲基噻唑连续化生产工艺
技术领域
1.本发明涉及化工合成技术领域,尤其涉及一种2
‑
氯
‑5‑
氯甲基噻唑连续化生产工艺。
背景技术:
[0002]2‑
氯
‑5‑
氯甲基噻唑,(2
‑
chloro
‑5‑
chloromethylthiazole),简称ccmt,分子式:c4h3cl2ns,cas号:105827
‑
91
‑
6,分子量:168,熔点:29.5
‑
30.0℃,工业品为无色至淡黄色液体或固体。它是合成农药噻虫嗪、噻虫胺、呋虫胺、医药利托那韦的重要中间体。以噻虫嗪、氯噻啉等为代表的噻唑类化合物,作为第二代烟碱类高效低毒杀虫剂,具有用量低、广谱、高效、与其它杀虫剂无交互抗性等特点,既对地上地下害虫有一定的防治性,又能对茎叶、土壤、种子进行处理,且在蔬菜中残留低、对环境污染少、对人类危害少,已成为目前农药开发的新热点。同时利托那韦作为人免疫缺陷病毒(hiv)的抑制剂不可或缺,其重要中间体2
‑
氯
‑5‑
氯甲基噻唑的需求量也在逐年增加。因此,2
‑
氯
‑5‑
氯甲基噻唑的研制和生产对于农药及医药的发展和创制具有极高的现实意义和应用前景。
[0003]
现阶段2
‑
氯
‑5‑
氯甲基噻唑主要的工业化合成工艺主要采用以反应釜间隔反应方式生产,操作人员多,劳动强度大,产能得不到最大化,且生产工艺安全系数不高,主要表现在以下方面:1)反应物料一次性加入容易导致氯化反应剧烈,而副产物如氯化氢气体易溶于体系中,氯化反应和脱除氯化氢气体的过程对压力的控制要求较高,容易引发安全事故;2)间隙通氯氯化的反应过程中,由于反应压力的变化,容易将反应物料倒吸至氯气系统,从而发生安全事故;3)间隙生产方式容易导致反应原料2
‑
氯
‑3‑
异硫氰
‑1‑
丙烯反应不完全,以及副产物增多,使得原材料消耗增大和成本升高的同时,后期除杂不易且工艺繁琐,增加了三废排放压力,也容易造成原料和副产物的残余量超标,进而使得产品具有质量不稳定、收率较低和不利于深加工等方面的缺陷;4)现有工艺得到的产品中,2
‑
氯
‑5‑
氯甲基噻唑含量通常为98
±
0.5%,副产物含量在1.0%以上,收率基本在75
‑
80%。
[0004]
因此,研究新的生产过程安全可控,反应过程质量稳定,能改善和提高产品质量、收率的连续化工业化生产工艺具有重要的现实意义。
技术实现要素:
[0005]
本发明提供一种2
‑
氯
‑5‑
氯甲基噻唑连续化生产工艺,用以解决现有工艺中由于反应釜间歇反应方式造成的生产工艺安全系数不高、设备利用率低、生产效率低下、产品质量不佳和收率低等问题,将2
‑
氯
‑5‑
氯甲基噻唑含量从现有工艺98%提高到99%以上,副产物含量从1.0%降低0.5%以下,收率从80%提高到91%以上,连续生产安全可控,副产物和废弃物生成量少,产品质量稳定,实现绿色环保、安全工业化生产。
[0006]
具体地,本发明提供的2
‑
氯
‑5‑
氯甲基噻唑连续化生产工艺,包括:
[0007]
提供有机溶剂,将原料2
‑
氯
‑3‑
异硫氰基
‑1‑
丙烯混入有机溶剂中形成混合液;
[0008]
提供连续氯化装置,在上述装置中通入氯气,进行氯化反应;
[0009]
提供精馏装置,在真空状态下依次进行溶剂蒸馏脱除、轻组份蒸馏脱除和脱重精馏,得到2
‑
氯
‑5‑
氯甲基噻唑产品;
[0010]
上述氯化反应采用连续进料连续溢流的方式进行;上述精馏装置采用连续进料连续减压精馏的操作条件。
[0011]
本发明中,通过连续化改进后,2
‑
氯
‑5‑
氯甲基噻唑的生产工艺简单可控,反应条件温和,体系能长期保持稳定,具有产品质量好、收率高的优点,达到了以高收率得到高纯度产品的目的,适合大规模工业化生产。
[0012]
本发明采用2
‑
氯
‑3‑
异硫氰基
‑1‑
丙烯和氯气进行氯化反应,能副产出洁净的盐酸,避免了使用磺酰氯副产二氧化硫气体和使用三氯氧磷副产含磷酸废水,既提高了产品纯度和反应收率,又减少了三废产生量,避免了排废压力和环境污染问题,具有环保性。
[0013]
进一步设置为,上述有机溶剂为乙腈、二氯乙烷、1,1,1
‑
三氯乙烷中的至少一种。
[0014]
进一步设置为,上述原料2
‑
氯
‑3‑
异硫氰基
‑1‑
丙烯与有机溶剂的重量比为1:1.5~3。
[0015]
进一步设置为,上述混合液的流量为1000~1400kg/h。
[0016]
进一步设置为,上述氯气的流量为550~600kg/h。
[0017]
进一步设置为,上述氯化反应的温度为25~40℃。
[0018]
进一步设置为,上述连续氯化装置还包括保温装置,上述保温装置的物料进料流量为1000~1400kg/h,温度为35~40℃。
[0019]
本发明通入氯气进行氯化反应得到产品,而非通过成盐操作得到产品的盐酸盐,避免了后期再加工提纯的工艺步骤和废弃物出产,符合绿色生产的理念。
[0020]
进一步设置为,溶剂蒸馏脱除的条件如下:温度为70~80℃、真空度为
‑
0.07~
‑
0.09mpa、薄膜蒸发器的蒸汽压力为0.15~0.18mpa。
[0021]
进一步设置为,轻组份蒸馏脱除的条件如下:温度为120~125℃、真空度为
‑
0.09~
‑
0.10mpa、薄膜蒸发器的蒸汽压力为0.18~0.23mpa。
[0022]
进一步设置为,脱重精馏的条件如下:温度为125~130℃、真空度为
‑
0.09~
‑
0.10mpa、薄膜蒸发器的蒸汽压力为0.20~0.30mpa。
[0023]
本发明通过在真空条件下进行连续性减压精馏,能降低产物沸点,从而在较低的温度下分离出产品,既能避免产物因高温分解或发生其它副反应,又能提高生产系统的安全性。
[0024]
本发明提供的连续化生产工艺,通过采用连续进料连续溢流、连续进料连续精馏的手段,实现如下的有益效果:
[0025]
1)本发明采用连续生产工艺,以2
‑
氯
‑3‑
异硫氰
‑1‑
丙烯在25~40℃范围内与氯气连续进料反应,反应条件温和可控,其连续反应过程中连续将氯化氢气体即时脱除,生产设备大幅度减少,该连续反应流程实现且适用于自动化仪表远程操作控制生产,替代间歇性釜式反应合成工艺,对产品质量、收率有明显改善与提高;
[0026]
2)本发明采用的是连续氯化反应器,连续定量进料,以溢流的方式采出物料,原料按规定比例稳定进料,氯化反应时间恒定,较大程度提高了反应传质效果,提高反应原料转化率和设备利用率,增加了产能和收率;
[0027]
3)本发明将气体氯化氢经溢流至脱氯化氢装置达到连续氯化反应与连续脱除氯
化氢的工艺要求,有利于提高生产及产品质量的稳定性;精馏通过连续脱溶、脱轻后连续精馏出高纯度产品,操作简单,非常适用于自动化生产控制;
[0028]
4)本发明合成2
‑
氯
‑5‑
氯甲基噻唑工艺氯化过程中动态维持整氯气用量,并通过连续溢流的方式带走氯化氢气体,大幅减少副反应产生,提高了原料利用率和产品质量,降低了三废处理难度和生产成本;
[0029]
5)本发明得到的2
‑
氯
‑5‑
氯甲基噻唑纯度从现有工艺98%提高到99%以上,副产物含量从1.0%降低至0.5%以下,氯化反应相比现有工艺收率由80%提高到91%以上,产品质量稳定。
附图说明
[0030]
为了更清楚地说明本发明实施例或现有技术中的技术方案,下面将对实施例或现有技术描述中所需要使用的附图作一简单地介绍,显而易见地,下面描述中的附图是本发明的一些实施例,对于本领域普通技术人员来讲,在不付出创造性劳动的前提下,还可以根据这些附图获得其他的附图。
[0031]
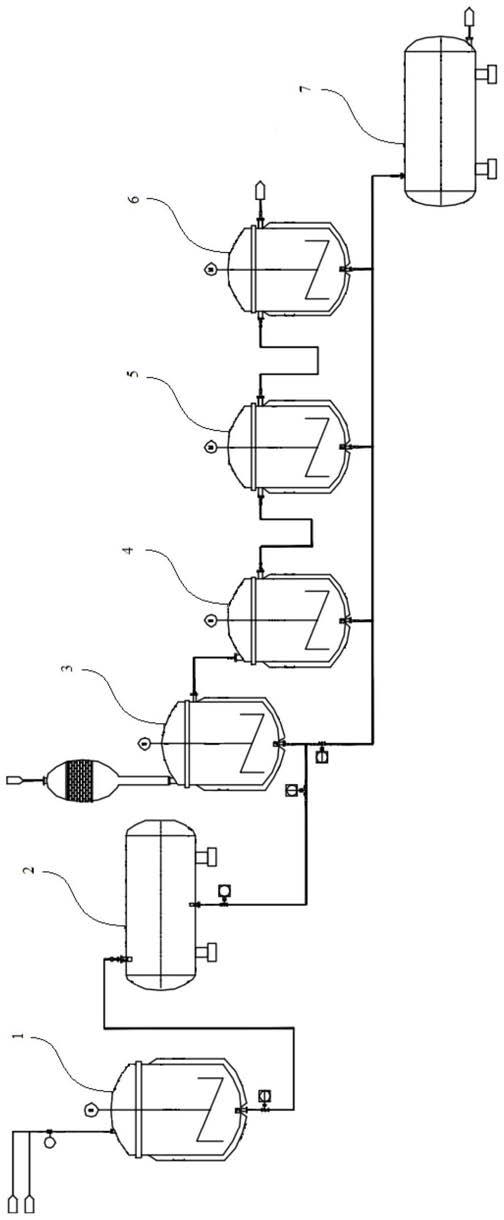
图1为本发明连续化生产工艺的流程及结构示意图。
[0032]
附图说明:1
‑
配置釜;2
‑
配置储罐;3
‑
氯化釜;4
‑
保温釜;5
‑
脱酸釜1;6
‑
脱酸釜2;7
‑
产物储罐。
具体实施方式
[0033]
为使本发明实施例的目的、技术方案和优点更加清楚,下面对本发明实施例中的技术方案进行清楚、完整地描述,显然,所描述的实施例是本发明一部分实施例,而不是全部的实施例。基于本发明中的实施例,本领域普通技术人员在没有作出创造性劳动前提下所获得的所有其它实施例,也属于本发明保护的范围。
[0034]
本发明中2
‑
氯
‑5‑
氯甲基噻唑的合成路线如下:
[0035]
1)脂化反应,方程式如下:
[0036]
ch2=ccl
‑
ch2cl+nascn
→
ch2=ccl
‑
ch2‑
c=n=s+nacl;
[0037]
2)氯化反应方程式
[0038]
ch2=ccl
‑
ch2‑
c=n=s+cl2→
c4h3cl2ns+hcl。
[0039]
在具体的实施方案中,氯气由氯气导管通入氯化反应装置内,氯气导管与氯化反应装置的溶液非接触设置或氯气导管上安装单向阀。
[0040]
在具体的实施方案中,氯化反应在氯化反应釜中进行,连续进料是将物料加进反应釜的底部,溢流出料是从设置在反应釜顶部的溢流口进行的。
[0041]
在具体的实施方案中,在多级连续氯化反应釜中,多组同时反应,连续进出料,生产效率提升,由于氯气不断被补充和溢流,能保证其充分反应和转化,同时避免了因间歇式反应釜中氯气过量而生成2
‑
氯
‑5‑
氯甲基噻唑盐酸盐,也避免了盐酸盐的碱洗等步骤以及相关设备投入,节约了生产成本,另外还降低了反应尾气的提纯和回收利用难度。
[0042]
在具体的实施方案中,氯化反应产生的气体氯化氢经导出,通入尾气系统进行回收利用,达到无有害废气排放的绿色环保生产目的。
[0043]
参见图1所示,本发明还提供了2
‑
氯
‑5‑
氯甲基噻唑连续化生产工艺及结构示意
图。具体的,2
‑
氯
‑
3异硫氰基
‑1‑
丙烯和有机溶剂从配置釜(1)顶部进料,经搅拌混合后,将形成得混合液送入配置储罐(2)中,然后在送入氯化釜(3)中,氯气从氯化釜(3)顶部送入,在氯化釜(3)中反应后,从釜上端溢流,进入保温釜(4)中,继续保温反应,然后溢流进入脱酸釜1(5)和脱酸釜2(6)中,脱除废气,产物进入产物储罐(7),后续进入连续精馏部分进行精制。
[0044]
需要理解的是,在本发明的附图1中,附图所示的方位或位置关系,仅是为了便于描述本发明的流程,而不是指示或暗示所指的装置或元件必须具有特定的方位、以特定的方位构造和操作,因此不能理解为对本发明的限制。
[0045]
更具体地,2
‑
氯
‑5‑
氯甲基噻唑连续化生产工艺,包括以下步骤:
[0046]
1)将2
‑
氯
‑
3异硫氰基
‑1‑
丙烯加入有机溶剂中,混合均匀,得到混合液备用;有机溶剂为乙腈、二氯乙烷、1,1,1
‑
三氯乙烷中的至少一种;2
‑
氯
‑
3异硫氰基
‑1‑
丙烯和有机溶剂的重量比为1:1.5~3;
[0047]
2)将上述混合液以1000~1400kg/h的流量送入氯化釜中,然后向其中以550~600kg/h的流量通入氯气,采用连续进料连续溢流的方式,在温度为25~40℃的条件下,进行氯化
‑
环合反应,然后从氯化釜溢流,以1000~1400kg/h的流量进入温度为35~40℃的保温釜中,连续氯化
‑
闭环得到含有2
‑
氯
‑5‑
氯甲基噻唑的混合溶液;上述溢流时通过脱酸釜1和2将反应产生的气体氯化氢导出,通入尾气系统进行回收利用;
[0048]
3)将上述含有2
‑
氯
‑5‑
氯甲基噻唑的混合溶液采用连续进料连续精馏的方式,在温度70~80℃、真空度
‑
0.07~
‑
0.09mpa、薄膜蒸发器的蒸汽压力为0.15~0.18mpa的条件下通过脱溶塔脱除溶剂,然后在温度120~125℃、真空度
‑
0.09~
‑
0.10mpa、薄膜蒸发器的蒸汽压力为0.18~0.23mpa的条件下通过脱轻塔脱除轻组份,然后在温度125~130℃、真空度
‑
0.09~
‑
0.10mpa、薄膜蒸发器的蒸汽压力为0.20~0.30mpa的条件下通过脱重塔将产品从塔中部精馏,得到2
‑
氯
‑5‑
氯甲基噻唑产品。
[0049]
实施例1:
[0050]2‑
氯
‑5‑
氯甲基噻唑连续化生产工艺,包括以下步骤:
[0051]
1)将200g的2
‑
氯
‑
3异硫氰基
‑1‑
丙烯加入360g的有机溶剂二氯乙烷中,混合均匀,得到混合液备用;
[0052]
2)将上述混合液以1000kg/h的流量送入氯化釜中,然后向上述氯化装置中以570kg/h的流量通入氯气,采用连续进料连续溢流的方式,在温度为30~35℃的条件下,进行氯化
‑
环合反应,然后从氯化釜溢流,以1000kg/h的流量进入温度为35℃的保温釜中,连续氯化
‑
闭环得到含有2
‑
氯
‑5‑
氯甲基噻唑的混合溶液;上述溢流时通过脱酸釜将反应产生的气体氯化氢导出,通入尾气系统进行回收利用;
[0053]
3)将上述含有2
‑
氯
‑5‑
氯甲基噻唑的混合溶液采用连续进料连续精馏的方式,在温度75~80℃、真空度
‑
0.085mpa、薄膜蒸发器的蒸汽压力为0.15mpa的条件下通过脱溶塔脱除溶剂,然后在温度120~125℃、真空度
‑
0.093mpa、薄膜蒸发器的蒸汽压力为0.19mpa的条件下通过脱轻塔脱除轻组份,然后在温度125~130℃、真空度
‑
0.093mpa、薄膜蒸发器的蒸汽压力为0.23mpa的条件下通过脱重塔将产品从塔中部精馏,得到2
‑
氯
‑5‑
氯甲基噻唑产品。
[0054]
该实施例中,精馏部分得到原料2
‑
氯
‑
3异硫氰基
‑1‑
丙烯79.7g,产品2
‑
氯
‑5‑
氯甲
基噻唑140.4g,产品含量99.5%,收率92.1%。
[0055]
实施例2:
[0056]2‑
氯
‑5‑
氯甲基噻唑连续化生产工艺,包括以下步骤:
[0057]
1)将120g的2
‑
氯
‑
3异硫氰基
‑1‑
丙烯加入250g的有机溶剂1,1,1
‑
三氯乙烷中,混合均匀,得到混合液备用;
[0058]
2)将上述混合液以1200kg/h的流量送入氯化釜中,然后向上述氯化装置中以600kg/h的流量通入氯气,采用连续进料连续溢流的方式,在温度为35~40℃的条件下,进行氯化
‑
环合反应,然后从氯化釜溢流,以1200kg/h的流量进入温度为40℃的保温釜中,连续氯化
‑
闭环得到含有2
‑
氯
‑5‑
氯甲基噻唑的混合溶液;上述溢流时通过脱酸釜将反应产生的气体氯化氢导出,通入尾气系统进行回收利用;
[0059]
3)将上述含有2
‑
氯
‑5‑
氯甲基噻唑的混合溶液采用连续进料连续精馏的方式,在温度73~78℃、真空度
‑
0.07mpa、薄膜蒸发器的蒸汽压力为0.18mpa的条件下通过脱溶塔脱除溶剂,然后在温度120~125℃、真空度
‑
0.097mpa、薄膜蒸发器的蒸汽压力为0.23mpa的条件下通过脱轻塔脱除轻组份,然后在温度125~130℃、真空度
‑
0.097mpa、薄膜蒸发器的蒸汽压力为0.28mpa的条件下通过脱重塔将产品从塔中部精馏,得到2
‑
氯
‑5‑
氯甲基噻唑产品。
[0060]
该实施例中,精馏部分得到原料2
‑
氯
‑
3异硫氰基
‑1‑
丙烯32.7g,产品2
‑
氯
‑5‑
氯甲基噻唑101.5g,产品含量99.6%,收率91.8%。
[0061]
实施例3:
[0062]2‑
氯
‑5‑
氯甲基噻唑连续化生产工艺,包括以下步骤:
[0063]
1)将150g的2
‑
氯
‑
3异硫氰基
‑1‑
丙烯加入300g的有机溶剂乙腈中,混合均匀,得到混合液备用;
[0064]
2)将上述混合液以1400kg/h的流量送入氯化釜中,然后向上述氯化装置中以560kg/h的流量通入氯气,采用连续进料连续溢流的方式,在温度为25~30℃的条件下,进行氯化
‑
环合反应,然后从氯化釜溢流,以1400kg/h的流量进入温度为38℃的保温釜中,连续氯化
‑
闭环得到含有2
‑
氯
‑5‑
氯甲基噻唑的混合溶液;上述溢流时通过脱酸釜将反应产生的气体氯化氢导出,通入尾气系统进行回收利用;
[0065]
3)将上述含有2
‑
氯
‑5‑
氯甲基噻唑的混合溶液采用连续进料连续精馏的方式,在温度70~75℃、真空度
‑
0.09mpa、薄膜蒸发器的蒸汽压力为0.17mpa的条件下通过脱溶塔脱除溶剂,然后在温度120~125℃、真空度
‑
0.095mpa、薄膜蒸发器的蒸汽压力为0.21mpa的条件下通过脱轻塔脱除轻组份,然后在温度125~130℃、真空度
‑
0.095mpa、薄膜蒸发器的蒸汽压力为0.24mpa的条件下通过脱重塔将产品从塔中部精馏,得到2
‑
氯
‑5‑
氯甲基噻唑产品。
[0066]
该实施例中,精馏部分得到原料2
‑
氯
‑
3异硫氰基
‑1‑
丙烯35.4g,产品2
‑
氯
‑5‑
氯甲基噻唑133.7g,产品含量99.7%,收率92.2%。
[0067]
实施例4:
[0068]2‑
氯
‑5‑
氯甲基噻唑连续化生产工艺,包括以下步骤:
[0069]
1)将80g的2
‑
氯
‑
3异硫氰基
‑1‑
丙烯加入150g的有机溶剂二氯乙烷中,混合均匀,得到混合液备用;
[0070]
2)将上述混合液以1200kg/h的流量送入氯化釜中,然后向上述氯化装置中以580kg/h的流量通入氯气,采用连续进料连续溢流的方式,在温度为30~35℃的条件下,进行氯化
‑
环合反应,然后从氯化釜溢流,以1200kg/h的流量进入温度为40℃的保温釜中,连续氯化
‑
闭环得到含有2
‑
氯
‑5‑
氯甲基噻唑的混合溶液;上述溢流时通过脱酸釜将反应产生的气体氯化氢导出,通入尾气系统进行回收利用;
[0071]
3)将上述含有2
‑
氯
‑5‑
氯甲基噻唑的混合溶液采用连续进料连续精馏的方式,在温度75~80℃、真空度
‑
0.08mpa、薄膜蒸发器的蒸汽压力为0.16mpa的条件下通过脱溶塔脱除溶剂,然后在温度120~125℃、真空度
‑
0.099mpa、薄膜蒸发器的蒸汽压力为0.20mpa的条件下通过脱轻塔脱除轻组份,然后在温度125~130℃、真空度
‑
0.099mpa、薄膜蒸发器的蒸汽压力为0.25mpa的条件下通过脱重塔将产品从塔中部精馏,得到2
‑
氯
‑5‑
氯甲基噻唑产品。
[0072]
该实施例中,精馏部分得到原料2
‑
氯
‑
3异硫氰基
‑1‑
丙烯12.3g,产品2
‑
氯
‑5‑
氯甲基噻唑94.3g,产品含量99.5%,收率91.0%。
[0073]
最后应说明的是,以上各实施例仅用以说明本发明的技术方案,而非对其限制;尽管参照前述各实施例对本发明进行了详细的说明,本领域的普通技术人员应当理解;其依然可以对前述各实施例所记载的技术方案进行修改,或者对其中部分或者全部技术特征进行等同替换;而这些修改或者替换,并不使相应技术方案的本质脱离本发明各实施例技术方案的范围。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1