山羊友好位点SETD5-IN和特异靶向该位点的sgRNA及其编码DNA和应用
山羊友好位点setd5
‑
in和特异靶向该位点的sgrna及其编码dna和应用
技术领域
1.本发明属于基因工程技术领域,具体涉及山羊友好位点setd5
‑
in和特异靶向该位点的sgrna及其编码dna和应用。
背景技术:
2.在生产转基因动物的过程中,将目的基因整合至动物基因组的特定位点(靶点)是其中的关键步骤。在选择靶点时,一方面应避免转入的片段影响动物的正常生理活动,因此,靶点通常位于基因组中的非编码区。rosa26(reverse orientation splice acceptorβ
‑
geo 26)基因是常见的外源基因插入位点,转录产物为长非编码rna(long none coding rna,lncrna),且可在全身各组织广泛表达,因此很多研究将rosa26基因视为友好的转基因位点。然而随着近年来人们对lncrna的深入研究,不断有报道证实,lncrna会参与细胞内多种过程的调控,非编码不等于没有生物学意义,只是以现有的检测技术较难捕获它们发挥功能的途径。此外,目前很多基因相关研究及产业应用往往需要时空特异性表达,而rosa26基因位点的启动子为全身广谱性表达,难以做到时空特异性表达。因此,新的基因编辑友好位点有待开发。
3.另一方面,基因编辑的效率也是选择靶点时需要考虑的因素。过去,研究者们仅将两端连接了靶点侧翼序列(同源臂)的目的基因转入细胞中,通过细胞自身的同源重组修复(homology
‑
directed repair,hdr)机制进行基因定点整合,然而这种方法不能高效地引起靶点处dna断裂,整合效率仅在0.01%左右,难以实现大规模应用。因此研究者们针对这一问题开发了可识别基因组特定靶点的人工核酸内切酶,可以更高效地诱导靶点处dna双链断裂(double strand break,dsb),当同时转入同源修复模板时,dsb可通过hdr途径修复,利用这一机制,可以更高效地将目的基因定点整合至靶点处。
4.近年常用的人工核酸内切酶主要有:zfn、talen、crispr/cas9系统等,特别是crispr/cas9系统,以其操作简单、成本低、效率高的特点,被广泛应用于多种动物的基因敲除和转基因动物生产。crispr/cas9系统包含cas9蛋白和小向导rna(small guide rna,sgrna),sgrna上有一段20nt的区域负责识别靶点。当基因组上存在crispr/cas9系统识别所必需的pam区(5
’‑
ngg),且pam区上游(5’端)的序列与sgrna匹配时,cas9蛋白就会切割基因组靶点部位dna双链,产生dsb。
5.然而,crispr/cas9系统也存在脱靶的问题。有研究发现,某些与sgrna识别区域并非完全匹配的序列也发生了dsb,这一情况被称为cas9的脱靶效应,脱靶效应会导致基因定点编辑的效率降低,同时可能使脱靶位点所在区域的功能受到影响。进一步研究发现,pam区前7~13nt的序列对sgrna引导的cas9蛋白的识别及切割起到较大的影响,这部分序列被称为种子序列,当种子序列能够完全与dna链匹配时就可能引导cas9蛋白进行切割。因此在筛选靶点时应使用种子序列+ngg与目的基因组进行比对,可以筛选出脱靶率较低的靶点,避免影响其他基因表达并提高基因定点整合效率。
6.因此,挖掘能够保证外源基因高效稳定表达、对转化体没有不利影响、可用于基因编辑的友好位点(安全位点),并筛选高效且特异性靶向该位点的crispr/cas9 sgrna序列,可为定点整合目的基因、生产转基因动物及相关研究提供有力的技术支撑,具有广泛的应用前景和重要的经济意义。
技术实现要素:
7.本发明的目的是提供一种可用于基因编辑的山羊友好位点setd5
‑
in,以及一种打靶效率高、脱靶率低的特异性靶向该友好位点的sgrna及其应用。所要解决的技术问题不限于如所描述的技术主题,本领域技术人员通过以下描述可以清楚地理解本文未提及的其它技术主题。
8.为实现上述目的,本发明首先提供了setd5
‑
in在作为山羊的基因编辑位点中的应用;所述setd5
‑
in是大小为2kb的双链dna分子,所述setd5
‑
in的核苷酸序列为seq id no.1。
9.所述应用的直接目的可以是疾病诊断目的和/或疾病治疗目的,也可以是非疾病诊断目的和/或非疾病治疗目的。
10.所述setd5
‑
in作为山羊的基因编辑位点即山羊友好位点setd5
‑
in。
11.所述友好位点是指基因组中外源基因定点整合的安全位点,整合在这个位点的基因能够稳定可靠的表达且不影响转入基因附近其他基因表达,而且能够适应不同类型的转基因表达盒的表达,对内源的基因结构和表达没有不利的影响。
12.本发明提供了山羊(gca_001704415.1_ars1)友好位点setd5
‑
in,该位点长度为2kb,位于山羊第22号染色体,setd5基因的第一内含子上(chr22:17026455
‑
17028455),同时距离rosa26基因第一外显子上游约2.4kb,序列如seq id no.1所示,其在基因组上的位置如图1所示。预测分析以及现有的注释均显示其中无特殊位点(如转录因子结合位点,可变剪接位点等)。因此,在该位点内进行基因编辑不会对setd5基因及邻近基因的表达产生影响。
13.本发明还提供了sgrna,所述sgrna特异性靶向于所述的setd5
‑
in。
14.进一步地,所述sgrna识别区的核苷酸序列为seq id no.2
‑
seq id no.7中的任一种。
15.所述sgrna特异靶向山羊友好位点setd5
‑
in。
16.其中,seq id no.2~4与seq id no.5~7分别靶向山羊友好位点setd5
‑
in(seq id no.1)的2条互补的dna双链。
17.本发明还提供了编码所述sgrna的dna分子。
18.进一步地,所述dna分子的核苷酸序列为seq id no.8
‑
seq id no.13中的任一种。
19.其中,seq id no.8为编码seq id no.2的dna序列,seq id no.9为编码seq id no.3的dna序列,seq id no.10为编码seq id no.4的dna序列,seq id no.11为编码seq id no.5的dna序列,seq id no.12为编码seq id no.6的dna序列,seq id no.13为编码seq id no.7的dna序列。
20.进一步地,sgrna靶点序列、sgrna识别区序列、转录(编码)sgrna序列的dna分子序列对应关系如下:
21.sgrna名称:setd5
‑
in
‑
sgrna2,靶点序列:actacacattatagatgact(seq id no.1的第868
‑
887),相应sgrna识别区序列如seq id no.2所示,相应sgrna识别区的编码dna序列如seq id no.8所示。
22.sgrna名称:setd5
‑
in
‑
sgrna3,靶点序列:taagtatcagatgtaactga(seq id no.1的第999
‑
1018),相应sgrna识别区序列如seq id no.3所示,相应sgrna识别区的编码dna序列如seq id no.9所示。
23.sgrna名称:setd5
‑
in
‑
sgrna4,靶点序列:gatgtaactgatggtatatt(seq id no.1的第1008
‑
1027),相应sgrna识别区序列如seq id no.4所示,相应sgrna识别区的编码dna序列如seq id no.10所示。
24.sgrna名称:setd5
‑
in
‑
sgrna5,靶点序列:caaagtatgtacatttctag(seq id no.1的第430
‑
449),识别该靶点的互补链序列(与seq id no.1的第430
‑
449位互补)的相应sgrna识别区序列如seq id no.5所示,相应sgrna识别区的编码dna序列如seq id no.11所示。
25.sgrna名称:setd5
‑
in
‑
sgrna6,靶点序列:tacttatctgatatggcatc(seq id no.1的第682
‑
701),识别该靶点的互补链序列(与seq id no.1的第682
‑
701位互补)的相应sgrna识别区序列如seq id no.6所示,相应sgrna识别区的编码dna序列如seq id no.12所示。
26.sgrna名称:setd5
‑
in
‑
sgrna7,靶点序列:caaacgccagagtcatacta(seq id no.1的第852
‑
871),识别该靶点的互补链序列(与seq id no.1的第852
‑
871位互补)的相应sgrna识别区序列如seq id no.7所示,相应sgrna识别区的编码dna序列如seq id no.13所示。
27.本发明还提供了dna分子,所述dna分子为所述的setd5
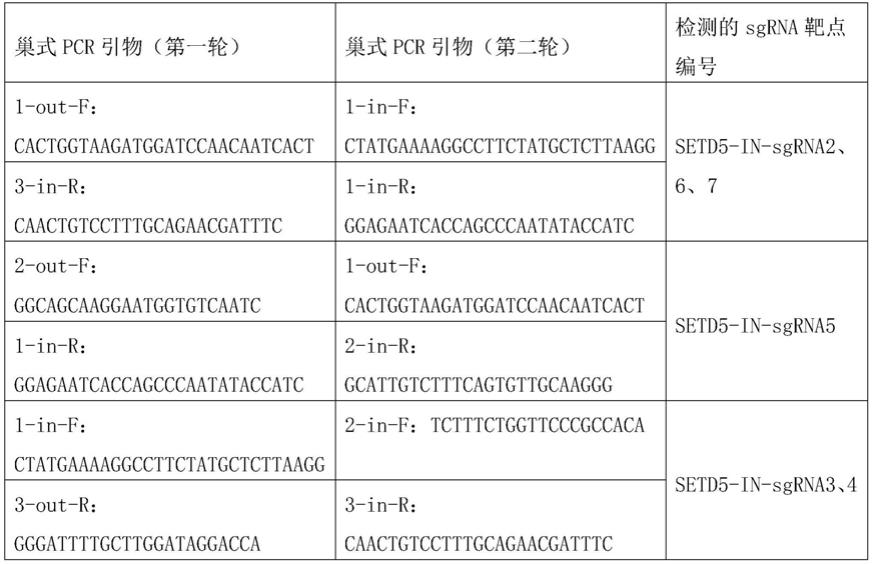
‑
in(seq id no.1)。
28.本发明还提供了生物材料,所述生物材料为下述b1)至b4)中的任一种:
29.b1)含有编码所述sgrna的dna分子的表达盒;
30.b2)含有编码所述sgrna的dna分子的重组载体、或含有b1)所述表达盒的重组载体;
31.b3)含有所述sgrna或含有编码所述sgrna的dna分子的重组微生物、或含有b1)所述表达盒的重组微生物、或含有b2)所述重组载体的重组微生物;
32.b4)含有所述sgrna或含有编码所述sgrna的dna分子的转基因动物细胞系、或含有b1)所述表达盒的转基因动物细胞系、或含有b2)所述重组载体的转基因动物细胞系。
33.上述生物材料中,所述载体可为质粒、黏粒、噬菌体或病毒载体。
34.上述生物材料中,所述微生物可为酵母、细菌、藻或真菌。其中,细菌可来自埃希氏菌属(escherichia),欧文氏菌(erwinia),根癌农杆菌属(agrobacterium)、黄杆菌属(flavobacterium),产碱菌属(alcaligenes),假单胞菌属(pseudomonas),芽胞杆菌属(bacillus)等。
35.上述生物材料中,所述重组载体具体可为crispr/cas9打靶载体。
36.含有本发明所述sgrna的crispr/cas9打靶载体属于本发明的保护范围。
37.上述生物材料中,所述crispr/cas9打靶载体可包括荧光筛选标记gfp。
38.进一步地,本发明提供了一种crispr/cas9打靶载体,可表达gfp荧光蛋白,进行基因编辑时,可通过流式筛选出转染了该打靶载体的细胞,提高阳性筛选效率。
39.本发明还提供了一种基因编辑的方法,所述方法包括将所述的setd5
‑
in作为山羊的基因编辑位点。
40.上述方法中,所述方法包括将编码所述sgrna的dna分子导入山羊细胞。
41.进一步地,所述方法包括将所述crispr/cas9打靶载体采用核转染的方式导入动物细胞。所述动物可为山羊。
42.本发明经过大量实验筛选发现,细胞转染cas9打靶载体时采用核转染的方式其转染效率优于脂质体转染和新型dna聚合转染试剂。因此优选采用核转染大分子量的打靶载体。
43.所述核转染是指综合应用传统的电穿孔技术及细胞特异性细胞核转染液,通过调整优化的电转参数(内存转染程序),直接把外源基因导入原代细胞及细胞系的细胞浆和细胞核中。
44.上述方法中,所述核转染采用v
‑
024电转染程序(简称v
‑
024程序,该程序是lonza电转仪的内置程序,仪器型号:lonza amaxa nucleofector 2b)。
45.优选地,在通过核转染向细胞转染cas9打靶载体时,可采用v
‑
024电转染程序,其效率高于n
‑
024电转染程序(简称n
‑
024程序,该程序是lonza电转仪的内置程序,仪器型号:lonza amaxa nucleofector 2b)。
46.本发明还提供了所述sgrna的下述任一种应用:
47.a1)在特异识别和/或靶向修饰山羊友好位点setd5
‑
in中的应用,
48.a2)在基因敲除、基因转入或定点过表达内源基因中的应用,
49.a3)在制备转基因动物中的应用,
50.a4)在制备转基因山羊中的应用,
51.a5)在利用相关时空特异性表达基因中的应用,
52.a6)在动物种质资源改良中的应用,
53.a7)在制备动物模型中的应用。
54.本发明还提供了所述dna分子,和/或,所述生物材料的下述任一应用:
55.a1)在特异识别和/或靶向修饰山羊友好位点setd5
‑
in中的应用,
56.a2)在基因敲除、基因转入或定点过表达内源基因中的应用,
57.a3)在制备转基因动物中的应用,
58.a4)在制备转基因山羊中的应用,
59.a5)在利用相关时空特异性表达基因中的应用,
60.a6)在动物种质资源改良中的应用,
61.a7)在制备动物模型中的应用。
62.本发明的有益效果在于:本发明提供了山羊友好位点setd5
‑
in,该位点可转入外源基因或定点过表达内源基因等,不会影响setd5基因及邻近基因的表达,为基因定点整合提供了新的安全的位点。同时,利用本发明提供的打靶效率高、脱靶率低的特异靶向上述友好位点的sgrna可以高效、特异地进行基因编辑,为研究家畜重要经济性状相关基因功能和生产转基因山羊奠定了基础。
附图说明
63.图1为友好位点setd5
‑
in在山羊基因组上的位置。
64.图2为山羊setd5内含子打靶示意图(以setd5
‑
in
‑
sgrna2和setd5
‑
in
‑
sgrna7为
例)。
65.图3为sgrna表达载体结构示意图。
66.图4为sgrna表达载体测序峰图结果。
67.图5为转染sgrna表达载体后培养3天的山羊成纤维细胞。左图为通过n
‑
024程序电转的山羊成纤维细胞;右图为通过v
‑
024程序电转的山羊成纤维细胞。
68.图6为t7e1酶切实验结果(通过n
‑
024程序转染)。
69.图7为t7e1酶切实验结果(通过v
‑
024程序转染)。
70.图8为双切型同源重组载体结构示意图。
71.图9为阳性细胞的pcr鉴定。
72.图10为阳性细胞的免疫荧光鉴定。
73.图11为通过wb对阳性细胞tlr2蛋白浓度的鉴定。
具体实施方式
74.下面结合具体实施方式对本发明进行进一步的详细描述,给出的实施例仅为了阐明本发明,而不是为了限制本发明的范围。以下提供的实施例可作为本技术领域普通技术人员进行进一步改进的指南,并不以任何方式构成对本发明的限制。
75.下述实施例中的实验方法,如无特殊说明,均为常规方法,按照本领域内的文献所描述的技术或条件或者按照产品说明书进行。下述实施例中所用的材料、试剂等,如无特殊说明,均可从商业途径得到。
76.下述实施例中的px458载体、pcdna3
‑
cd68 promoter/enhancer载体、prosa26
‑
promoter载体购于addgene公司;pcmv
‑
tdtomato载体购于clontech公司;限制性核酸内切酶、t7e1酶、t4连接酶及buffer购于neb公司;引物合成及序列测定由苏州金唯智公司完成;kod plus neo高保真pcr酶购于东洋纺公司;细胞电转液s1、s2、电转杯购于lonza公司;无缝克隆试剂盒geneart gibson assembly购于thermo fisher公司。
77.实施例1、山羊友好位点setd5
‑
in的确定
78.所述山羊友好位点setd5
‑
in长度为2kb,位于山羊第22号染色体,在山羊基因组(gca_001704415.1)上的位置是chr22:17026455
‑
17028455,该区域位于setd5基因的第一内含子上,序列如seq id no.1所示,其在基因组上的位置如图1所示。基于animaltfdb数据库的预测显示该友好位点中无转录因子结合位点;基于ncbi现有山羊基因组的*.gff注释文件记录的基因位置信息,显示该友好位点中无可变剪接位点。在该位点内进行基因编辑不会对setd5基因及邻近基因的表达产生影响。
79.实施例2、crispr/cas9打靶载体的构建
80.crispr/cas9打靶载体(sgrna表达载体)的结构如图3所示。
81.1)根据crispr/cas9系统的sgrna筛选原则,筛选山羊setd5
‑
in位点区域内的pam序列(5
’‑
ngg)及对应的sgrna靶点,并进行脱靶效率预测:选取pam序列5’端的13nt序列作为种子序列,与ncbi在线数据库进行比对。使用sgrna结构在线预测网站(http://unafold.rna.albany.edu/q=mfold/rna
‑
folding
‑
form/)评估各靶点对应的sgrna的结构及自由能,确认sgrna上的识别序列不与自身配对。
82.sgrna靶点序列(不含pam区,下表中序列为靶点区域基因组dna正链序列)及相应
sgrna识别区序列、转录(编码)sgrna序列的dna分子的序列对应关系如下:
83.sgrna名称:setd5
‑
in
‑
sgrna2,靶点序列:actacacattatagatgact(seq id no.1的第868
‑
887),相应sgrna识别区序列如seq id no.2所示,相应sgrna识别区的编码dna序列如seq id no.8所示。
84.sgrna名称:setd5
‑
in
‑
sgrna3,靶点序列:taagtatcagatgtaactga(seq id no.1的第999
‑
1018),相应sgrna识别区序列如seq id no.3所示,相应sgrna识别区的编码dna序列如seq id no.9所示。
85.sgrna名称:setd5
‑
in
‑
sgrna4,靶点序列:gatgtaactgatggtatatt(seq id no.1的第1008
‑
1027),相应sgrna识别区序列如seq id no.4所示,相应sgrna识别区的编码dna序列如seq id no.10所示。
86.sgrna名称:setd5
‑
in
‑
sgrna5,靶点序列:caaagtatgtacatttctag(seq id no.1的第430
‑
449),识别该靶点的互补链序列(与seq id no.1的第430
‑
449位互补)的相应sgrna识别区序列如seq id no.5所示,相应sgrna识别区的编码dna序列如seq id no.11所示。
87.sgrna名称:setd5
‑
in
‑
sgrna6,靶点序列:tacttatctgatatggcatc(seq id no.1的第682
‑
701),识别该靶点的互补链序列(与seq id no.1的第682
‑
701位互补)的相应sgrna识别区序列如seq id no.6所示,相应sgrna识别区的编码dna序列如seq id no.12所示。
88.sgrna名称:setd5
‑
in
‑
sgrna7,靶点序列:caaacgccagagtcatacta(seq id no.1的第852
‑
871),识别该靶点的互补链序列(与seq id no.1的第852
‑
871位互补)的相应sgrna识别区序列如seq id no.7所示,相应sgrna识别区的编码dna序列如seq id no.13所示。
89.2)根据上述6个靶点序列,设计相应引物,具体序列如表1。
90.表1针对setd5
‑
in不同靶点的引物
91.引物核苷酸名称序列(5
’‑3’
)setd5
‑
in
‑
sgrna2
‑
fcaccgactacacattatagatgactsetd5
‑
in
‑
sgrna2
‑
raaacagtcatctataatgtgtagtcsetd5
‑
in
‑
sgrna3
‑
fcaccgtaagtatcagatgtaactgasetd5
‑
in
‑
sgrna3
‑
raaactcagttacatctgatacttacsetd5
‑
in
‑
sgrna4
‑
fcaccgatgtaactgatggtatattsetd5
‑
in
‑
sgrna4
‑
raaacaatataccatcagttacatcsetd5
‑
in
‑
sgrna5
‑
fcaccgctagaaatgtacatactttgsetd5
‑
in
‑
sgrna5
‑
raaaccaaagtatgtacatttctagcsetd5
‑
in
‑
sgrna6
‑
fcaccgatgccatatcagataagtasetd5
‑
in
‑
sgrna6
‑
raaactacttatctgatatggcatcsetd5
‑
in
‑
sgrna7
‑
fcaccgtagtatgactctggcgtttgsetd5
‑
in
‑
sgrna7
‑
raaaccaaacgccagagtcatactac
92.3)将合成的上述引物均用ddh2o稀释成100μm的溶液,在每个sgrna退火体系中分别添加相应的f和r引物溶液各1μl,t4连接酶buffer 1μl,ddh2o 7μl,混匀。将上述退火体系放入pcr仪中:95℃,2min;以
‑
5℃/s的速度降至25℃,随后置于冰上,令sgrna退火体系中的f和r引物互补配对,形成含有黏性末端的双链寡核苷酸(oligo);并取1μl oligo,用ddh2o稀释100倍,得到oligo稀释液。
93.4)将px458载体用限制性内切酶bbsi酶切并回收纯化,得到酶切后的线性骨架载体,并在每个连接体系中添加100ng酶切后的线性骨架载体;在每个连接体系中加入1μl步骤3)获得的oligo稀释液、1μl t4连接酶buffer、1μl t4连接酶,并用ddh2o补足至10μl,混匀,置于16℃过夜反应。通过常规转化法进行转化、涂板。待单菌落长成后,挑取数个扩大培养并测序验证(测序结果见图4,加深标识的部分为sgrna序列信息及其测序峰图结果),比对结果显示sgrna识别区的编码dna已成功连入骨架载体,针对6个靶点的crispr/cas9打靶载体(sgrna表达载体)均成功构建,分别命名为:setd5
‑
in
‑
sgrna2表达载体、setd5
‑
in
‑
sgrna3表达载体、setd5
‑
in
‑
sgrna4表达载体、setd5
‑
in
‑
sgrna5表达载体、setd5
‑
in
‑
sgrna6表达载体和setd5
‑
in
‑
sgrna7表达载体。
94.实施例3、细胞水平的基因编辑效率验证
95.步骤1)制备山羊成纤维细胞。在山羊妊娠40d后剖腹产取出胎儿,去除头,尾,四肢与内脏后,将组织剪成大小为1mm3的小块。将剪碎的组织块均匀放置在培养皿中,将培养皿倒置在38.5℃、5%co2、饱和湿度的细胞培养箱中培养2h使组织块贴附于培养皿表面。加入少量含10%fbs的dmem/f12培养液,继续在培养箱中培养24h,加入3ml培养基继续培养。以后每3d换液一次,每天观察,记录细胞的生长状况。
96.步骤2)sgrna表达载体(crispr/cas9打靶载体)去内毒素大提。
97.按照无内毒素质粒大提试剂盒(天根生化科技有限公司)上提供的方法,提取sgrna表达载体(crispr/cas9打靶载体)质粒,所提的质粒用于细胞转染。
98.步骤3)细胞转染
99.本发明经过大量实验筛选发现,细胞转染cas9打靶载体时采用核转染的方式其转染效率优于脂质体转染和新型dna聚合转染试剂。因此优选采用核转染大分子量的crispr/cas9打靶载体。所述核转染是指综合应用传统的电穿孔技术及细胞特异性细胞核转染液,通过调整优化的电转参数(内存转染程序),直接把外源基因导入原代细胞及细胞系的细胞浆和细胞核中。
100.采用amaxa’s nucleofector电转仪(lonza)进行电转,具体流程如下:
101.①
配制电转液:将s1与s2溶液以1:50的体积比混合均匀。
102.②
消化山羊成纤维细胞,用pbs将贴壁细胞吹打下来并转移至1.5ml离心管(每管约2
×
106个细胞),1500rpm/min,离心5min。
103.③
尽量去除上清液。用100μl电转液悬浮细胞。
104.④
向细胞悬液中加入4μg待转染的sgrna表达载体,混和均匀后加入电转杯的电极之间,注意避免产生气泡。
105.⑤
用n
‑
024程序电转山羊成纤维细胞,向电转后的细胞中立即加入500μldmem:f12培养基,静置10min。
106.⑥
将电转后的细胞铺至6孔板,用含20%fbs的培养基培养。
107.⑦
待转染后的细胞贴壁后,更换为含15%fbs的培养基培养。
108.同时,为验证不同电转程序的细胞转染效果,另准备1组细胞转染体系,将setd5
‑
in
‑
sgrna2表达载体通过v
‑
024程序(该程序是lonza电转仪的内置程序,仪器型号:lonza amaxa nucleofector 2b)转染入山羊成纤维细胞(除电转程序外,其他实验操作同上)。转染后的细胞培养3d,观察gfp(绿色荧光蛋白)的表达情况,结果如图5所示。
109.步骤4)t7e1酶切检测基因编辑效率
110.将步骤3)中转染后培养3d的山羊成纤维细胞消化并提取基因组dna,利用相应的t7e1检测引物对sgrna靶点周围约500bp的区域进行巢式pcr扩增,两轮扩增pcr体系及过程如下:
111.第一轮pcr体系:基因组dna——100ng,引物(10μm)——各0.75μl,dntp(2mm)——2.5μl,10
×
buffer——2.5μl,mgso4(2mm)——1.5μl,kod plus neo——0.5μl,ddh2o——稀释至25μl;反应条件:94℃2min;98℃10s,62℃30s,68℃40s,20个循环;68℃5min;4℃,∞。
112.第二轮pcr体系:第一轮pcr产物——1μl,引物(10μm)——各1.5μl,dntp(2mm)——5μl,10
×
buffer——5μl,mgso4(2mm)——3μl,kod plus neo——1μl,ddh2o——稀释至50μl;反应条件:94℃2min;98℃10s,62℃30s,68℃18s,35个循环;68℃5min;4℃,∞。
113.检测各sgrna靶点对应的所用的巢式引物名称及序列如表2所示:
114.表2巢式pcr引物列表
[0115][0116]
取5μl上述pcr体系加入新的pcr管中,加入1.1μl neb 2buffer、4.4μl ddh2o,混合均匀。通过以下程序进行退火杂交:95℃,10min;以
‑
2℃/s的速率降至85℃;以
‑
0.1℃/s的速率降至25℃。
[0117]
在杂交体系中加入0.5μl t7e1酶,混匀后37℃反应30min。加入2μl 6
×
loading buffer,通过凝胶电泳检测基因编辑效率。
[0118]
t7e1酶切结果如图6(n
‑
024程序转染)、图7(v
‑
024程序转染)所示,图中空心箭头指示条带为未被t7e1酶切的条带,实心箭头指示条带为t7e1酶切产物。通过灰度值分析软件计算各靶点的基因编辑效率(indel%)如表3所示,说明上述6个靶点均可介导基因编辑,且v
‑
024程序转染效率优于n
‑
024。
[0119]
表3基因编辑效率(indel%)
[0120][0121]
其中,fcut=(b+c)/(a+b+c)
[0122]
a为未被t7e1酶切的条带大小,b、c为t7e1酶切产物大小
[0123]
实施例4、tlr2基因定点整合setd5
‑
in载体构建
[0124]
实施例4用于说明针对本发明所述友好位点setd5
‑
in的同源重组载体。为提高定点整合效率,同时在同源左臂的上游及同源右臂的下游分别引入了setd5
‑
in
‑
sgrna2位点所在的靶点识别序列,可构建双切型同源重组载体。
[0125]
1)同源臂的构建
[0126]
以山羊基因组dna为模板,ha
‑
l
‑
f、ha
‑
l
‑
r为引物进行pcr,获得同源左臂;以山羊基因组dna为模板,ha
‑
r
‑
f、ha
‑
r
‑
r为引物进行pcr,获得同源右臂;
[0127]
ha
‑
l
‑
f:cccaagctatggggtatctactcaattca
[0128]
ha
‑
l
‑
r:atctataatgtgtagtatgactctggcgtt
[0129]
ha
‑
r
‑
f:gggttcatttacacaaaaccgaacatac
[0130]
ha
‑
r
‑
r:ttaatgctgttggctgcagg
[0131]
2)添加友好位点setd5
‑
in
‑
sgrna2所在的靶点识别序列
[0132]
在同源左臂的上游通过pcr添加如下序列(5
’‑3’
):
[0133]
actacacattatagatgactggg,获得上游含有setd5
‑
in
‑
sgrna2位点所在靶点识别序列的同源臂left(ha
‑
l);
[0134]
在同源右臂的下游通过pcr添加如下序列(5
’‑3’
):
[0135]
cccagtcatctataatgtgtagt,获得下游含有setd5
‑
in
‑
sgrna2位点所在靶点识别序列的同源臂right(ha
‑
r);
[0136]
3)将待转入的目的基因、poly(a)、同源臂ha
‑
l和ha
‑
r、cmv启动子、tdtomato报告基因序列连接至骨架载体prosa26
‑
promoter,构建针对友好位点setd5
‑
in
‑
sgrna2的双切型同源重组载体。具体方法如下:
[0137]
poly(a)利用引物pa
‑
f(ctgtgccttctagttgccag)、pa
‑
r(ccatagagcccaccgcatcc)扩增自pcdna3
‑
cd68 promoter/enhancer载体,使用无缝克隆试剂盒将poly(a)组装至使用限制性内切酶bamhi
‑
hf、xbai酶切的prosa26
‑
promoter载体;利用xbai和saci
‑
hf酶切上述载体,通过无缝克隆将ha
‑
r添加至上述构建载体poly(a)元件3’端;获得的载体使用acc651和sali酶切,并通过无缝克隆将ha
‑
l组装至上述构建载体rosa26
‑
promoter元件5’端;将获
得的载体使用ecori
‑
hf和pmli酶切;通过常规方法提取山羊血液总rna,并反转录获得cdna,以此为pcr模板,使用引物tlr2
‑
u(gcctctgatcaggcttcttc)、tlr2
‑
l(cctaggaccttattgcagct)扩增目的基因片段(tlr2基因外显子区);将获得的目的基因片段通过无缝克隆组装至上述酶切后的载体;最后,将获得的载体使用saci
‑
hf酶切,通过无缝克隆在ha
‑
r下游嵌入cmv启动子及tdtomato报告基因序列(cmv
‑
tdtomato),获得针对友好位点setd5
‑
in
‑
sgrna2的双切型同源重组载体。上述cmv
‑
tdtomato片段使用引物td
‑
u、td
‑
l扩增自pcmv
‑
tdtomato载体。
[0138]
双切型同源重组载体的结构如图8所示,其中双侧同源臂外侧的红色箭头处为setd5
‑
in
‑
sgrna2位点所在的靶点识别序列。
[0139]
实施例5、tlr2基因定点整合setd5
‑
in及表达
[0140]
实施例5用于说明利用实施例2所述打靶载体与实施例4所述的同源重组载体在转基因方面的应用。
[0141]
1、阳性单克隆细胞筛选
[0142]
1)山羊成纤维细胞在含10%fbs dmem的100mm细胞培养皿中培养,待细胞长满后用0.25%的胰蛋白酶消化细胞(约4
×
106个细胞),用pbs将贴壁细胞吹打下来并转移至1.5ml离心管,1500rpm/min,离心5min。
[0143]
2)尽量去除上清液,用100μl电转液悬浮细胞,并向悬浮液中加入5μg质粒(实施例2中的crispr/cas9打靶载体(sgrna表达载体)setd5
‑
in
‑
sgrna2表达载体2.5μg,实施例4中的双切型同源重组载体2.5μg),混合均匀后加入电转杯。
[0144]
3)使用v
‑
024程序电转山羊成纤维细胞,电转后的细胞中立即加入500μldmem:f12培养基,静置10min;将电转后的细胞铺至6孔板,用含20%fbs的培养基培养;待转染后的细胞贴壁后,更换为含15%fbs的培养基培养。
[0145]
4)转染的细胞培养3d后,将其消化并转移至流式管中,通过流式分选仪筛选同时表达gfp及tdtomato的细胞。将分选获得的细胞以500个细胞/100mm培养皿的密度铺板,使用含20%fbs的dmem:f12进行培养。培养10~14d时使用克隆环挑取细胞单克隆至96孔板中培养。
[0146]
5)待96孔板中的细胞单克隆汇合度达到约100%时,消化并取70%的细胞转移至新孔,标记对应原孔的编号,原孔中的剩余细胞继续培养;其中新孔中的细胞用于阳性细胞单克隆的鉴定,原孔中的细胞用于后续鉴定阳性细胞单克隆的扩大培养。
[0147]
2、阳性细胞单克隆的pcr鉴定
[0148]
1)待新孔中的细胞密度达到90%时,消化细胞至1.5ml离心管中。12000rpm/min,离心3min,弃去上清,加入50μl的裂解液,充分悬浮细胞,按以下程序裂解:65℃,30min;95℃,15min;16℃,∞。
[0149]
2)利用如下引物(5
’‑3’
)进行阳性细胞单克隆的pcr鉴定
[0150]
r
‑
u:ttgcttggtaagcgcggatat
[0151]
r
‑
l:agagacaccgaaccacacga
[0152]
pcr反应体系如下:步骤1)获得的细胞裂解液1μl作为模板,引物(10μm)各0.5μl,ddh2o:10.5μl,max pcr master mix:12.5μl。pcr反应条件为:94℃2min;98℃10s,62℃30s,72℃1min30s,35个循环;72℃5min;4℃,∞。扩增完成后,用1.5%的琼脂糖
凝胶电泳观察结果。
[0153]
进行阴性对照实验时,使用野生型山羊成纤维细胞基因组dna作为模板。引物r
‑
u位于左同源臂ha
‑
l的上游,引物r
‑
l则位于敲入片段mrosa26启动子上,因此发生了同源重组的阳性细胞基因组能扩增出1420bp的片段,而未发生同源重组的细胞基因组则不能扩增出1420bp的片段,扩增鉴定结果如图9所示。其中,m3为marker3,control为野生型山羊成纤维细胞基因组dna扩增结果,白色箭头指示为阳性细胞(转基因阳性细胞,以“myc
‑
tlr2”表示)扩增结果。说明外源基因在目的位点setd5
‑
in处发生了定点整合。
[0154]
3、细胞水平免疫荧光和wb实验过程和结果
[0155]
1)山羊成纤维细胞在含10%fbs的dmem培养基中培养,将野生型山羊成纤维细胞(wt)和转基因阳性细胞克隆以2
×
104个细胞/孔的密度铺在腔式细胞载玻片上,待细胞密度达到80%,吸去上清,向孔中加入100μl冰甲醇,4℃孵育20min。弃去甲醇,加入免疫荧光封闭液,室温孵育1h。弃去封闭液,使用免疫荧光一抗稀释液按比例稀释myc标签抗体,每孔加入100μl稀释后的抗体,4℃孵育过夜。弃去一抗,使用无菌pbs洗涤3次,按1:500的比例使用免疫荧光二抗稀释液稀释alexa flour 594标记的二抗,每孔加入100μl,室温孵育1h。弃去二抗,使用无菌pbs洗涤3次,加入dapi染核工作液,室温孵育10min。吸弃dapi,无菌pbs洗涤2次。拆下载玻片,滴加抗荧光淬灭剂,盖上盖玻片,使用激光共聚焦显微镜观察处理后的细胞。结果如图10所示,阳性细胞(图中为“myc
‑
tlr2”)能与myc标签一抗结合显示红色荧光,wt细胞因为没有myc标签而未被红色标记。
[0156]
2)将阳性转基因细胞以1
×
106的密度铺在100mm细胞培养皿中,在37℃细胞培养箱中过夜培养,吸去上清,无菌pbs清洗细胞2次。消化细胞,用pbs重悬细胞,以2000rpm/min转速离心5min。吸弃上清,用200μl加入了pmsf的ripa细胞裂解液,用移液器吹匀。将裂解液冰浴孵育30min,以2000rpm/min转速离心10min,留上清弃沉淀,该上清为转基因细胞蛋白样品。用bca法检测样品浓度,加入蛋白loading buffer(蛋白样与myc标准样),沸水浴5min。向蛋白胶中每孔加入转基因细胞蛋白样品与myc标准样(myc标签重组蛋白,购自proteintech公司),80v跑胶30min,转为120v继续跑胶1h。320ma转膜1h,用兔源myc标签抗体作为一抗,羊抗兔抗体作为二抗。在膜上滴加ecl发光液,wb仪器检测条带,使用image j软件检测灰度值。根据标准样的灰度值制作标准曲线,计算转基因细胞样品的tlr2浓度。结果如图11所示,转基因细胞样品(图中为“myc
‑
tlr2”)的tlr2浓度为81ng/mg。
[0157]
以上对本发明进行了详述。对于本领域技术人员来说,在不脱离本发明的宗旨和范围,以及无需进行不必要的实验情况下,可在等同参数、浓度和条件下,在较宽范围内实施本发明。虽然本发明给出了特殊的实施例,应该理解为,可以对本发明作进一步的改进。总之,按本发明的原理,本技术欲包括任何变更、用途或对本发明的改进,包括脱离了本技术中已公开范围,而用本领域已知的常规技术进行的改变。按以下附带的权利要求的范围,可以进行一些基本特征的应用。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1