氢化氧杂蒽酮衍生物及其制备方法和应用
1.本发明属于生物医药技术领域,特别涉及氢化氧杂蒽酮衍生物及其制备方法和应用。
背景技术:
2.氧杂蒽酮是一种异三环化合物,其衍生物广泛存在于自然界中,其中包括许多芳环被氢化或高度氧化的类似物。这些化合物因取代基不同而表现出不同的生物活性,包括调节机体免疫、抗肿瘤以及抗菌抗炎等,是研发新型抗生素的一类重要分子结构。天然来源的氧杂蒽酮类化合物往往具有更加复杂的化学结构和更加多样的官能团取代,从而表现出更显著的生物活性,尤其是从微生物资源中获得的氧杂蒽酮,是发现天然来源药物先导化合物的潜力分子。
技术实现要素:
3.本发明的目的在于提供氢化氧杂蒽酮衍生物,该系列氢化氧杂蒽酮衍生物能够抗植物病原菌并具备抗氧化活性,可用于制备农用抗生素和抗氧化药物。
4.本发明还提供了氢化氧杂蒽酮衍生物的制备方法和应用。
5.本发明通过以下技术方案实现:
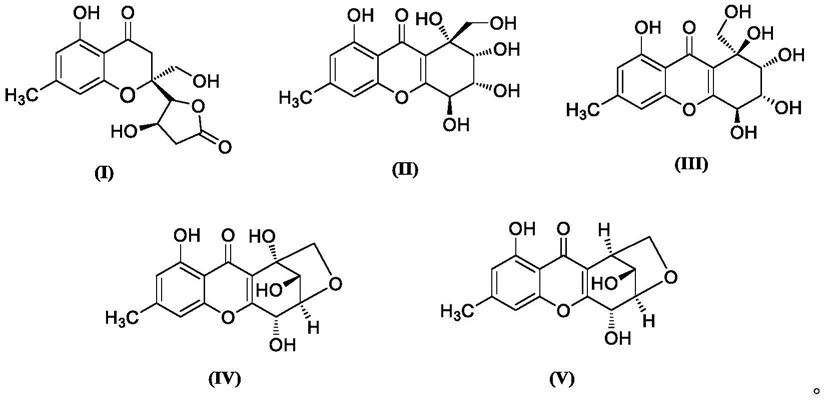
6.本发明提供氢化氧杂蒽酮衍生物,所述氢化氧杂蒽酮衍生物包括phomops i sxanthones h~l中的至少一种,phomops i sxanthones h~l的结构式分别如(i)~(v)所示:
[0007][0008]
基于同一发明构思,本发明提供一种氢化氧杂蒽酮衍生物的制备方法,所述制备方法包括:
[0009]
将拟茎点霉菌在添加有丁酸钠的培养基中进行发酵,发酵结束后收集发酵液和菌丝体;
[0010]
将所述发酵液进行萃取、将所述菌丝体进行浸提,合并提取液,后挥发溶剂,获得
发酵粗提物;
[0011]
将所述发酵粗提物进行色谱分离,洗脱剂采用氯仿
‑
甲醇体系,收集粗组分fr.1~fr.3;将粗组分fr.1进行色谱分离,洗脱剂采用氯仿
‑
甲醇体系,获得化合物phomopsisxanthone h;
[0012]
将粗组分fr.2进行色谱分离,洗脱剂采用石油醚
‑
乙酸乙酯体系,得到化合物phomopsisxanthone i;
[0013]
将粗组分fr.3进行色谱分离,洗脱剂采用甲醇
‑
水体系,得到化合物phomopsisxanthone j~l。
[0014]
进一步的,所述拟茎点霉菌为phomopsis sp.ye3350,菌株phomopsis sp.ye3350已于2020年8月3日保藏于中国典型培养物保藏中心(简称为cctcc),保藏编号为cctcc no.m 2020394。保藏地址为中国,武汉,武汉大学,分类学命名为phomopsis sp.。
[0015]
进一步的,所述将拟茎点霉菌在添加有丁酸钠的培养基中进行发酵,发酵结束后收集发酵液和菌丝体,具体包括:
[0016]
将拟茎点霉菌在添加有丁酸钠的pdb培养基中进行发酵,发酵结束后收集发酵液和菌丝体;
[0017]
所述添加有丁酸钠的pdb培养基通过以下方法制备:
[0018]
取200~400份去皮马铃薯切块,加入1000份水煮沸后过滤,获得马铃薯滤液;
[0019]
向所述马铃薯滤液中加入葡萄糖20~50份,磷酸氢二钾3~5份,硫酸镁3~5份和维生素b
1 0.01~0.03份,获得pdb培养基;
[0020]
向所述pdb培养基中加入丁酸钠,获得所述添加有丁酸钠的pdb培养基;
[0021]
所述pdb培养基中,马铃薯浓度为0.2~0.4g/ml,葡萄糖浓度为0.02~0.05g/ml;所述添加有丁酸钠的pdb培养基中,所述丁酸钠浓度为200~500μm。
[0022]
进一步的,所述将所述发酵液进行萃取、将所述菌丝体进行浸提,合并提取液,后挥发溶剂,获得发酵粗提物,具体包括:
[0023]
采用乙酸乙酯萃取所述发酵液、采用甲醇浸提所述菌丝体,合并提取液,后挥发溶剂,获得发酵粗提物。
[0024]
进一步的,所述将所述发酵粗提物进行色谱分离,洗脱剂采用氯仿
‑
甲醇体系,收集粗组分fr.1~fr.3,具体包括:
[0025]
将所述发酵粗提物进行色谱分离,洗脱剂采用氯仿
‑
甲醇体系,氯仿和甲醇从体积比10:1至1:1对所述发酵粗提物进行梯度洗脱,收集氯仿和甲醇体积比为9:1时洗脱得到的粗组分fr.1,收集氯仿和甲醇体积比为8:2时洗脱得到的粗组分fr.2,收集氯仿和甲醇体积比为7:3时洗脱得到的粗组分fr.3。
[0026]
进一步的,所述将粗组分fr.1进行色谱分离,洗脱剂采用氯仿
‑
甲醇体系,获得化合物phomopsisxanthone h,具体包括:
[0027]
将粗组分fr.1进行色谱分离,洗脱剂采用氯仿
‑
甲醇体系,氯仿和甲醇从体积比15:1至7:3对所述粗组分fr.1进行梯度洗脱,收集氯仿和甲醇体积比为9:1时洗脱得到的馏分,馏分在丙酮和甲醇体积比为3:1的混合溶剂中进行重结晶,获得化合物phomopsisxanthone h。
[0028]
进一步的,所述将粗组分fr.2进行色谱分离,洗脱剂采用石油醚
‑
乙酸乙酯体系,
得到化合物phomopsisxanthone i,具体包括:
[0029]
将粗组分fr.2进行色谱分离,洗脱剂采用石油醚
‑
乙酸乙酯体系,石油醚和乙酸乙酯从体积比9:1至7:3对所述粗组分fr.2进行梯度洗脱,收集石油醚和乙酸乙酯体积比为9:1时洗脱得到的馏分,馏分在甲醇和水体积比为8:2的混合溶剂中进行重结晶,获得化合物phomopsisxanthone i。
[0030]
进一步的,所述将粗组分fr.3进行色谱分离,洗脱剂采用甲醇
‑
水体系,得到化合物phomopsisxanthone j~l,具体包括:
[0031]
将粗组分fr.3进行色谱分离,洗脱剂采用甲醇
‑
水体系,甲醇和水从体积比1:1至9:1对所述粗组分fr.3进行梯度洗脱,收集甲醇和水体积比为6:4时洗脱得到的馏分,挥发洗脱剂,获得化合物phomopsisxanthone j;收集甲醇和水体积比为7:3时洗脱得到的馏分,挥发洗脱剂,获得化合物phomopsisxanthone k;收集甲醇和水体积比为8:2时洗脱得到的馏分,挥发洗脱剂,获得化合物phomopsisxanthone l。
[0032]
基于同一发明构思,本发明提供氢化氧杂蒽酮衍生物在制备农用抗生素和抗氧化药物中的应用。
[0033]
本发明实施例中的一个或多个技术方案,至少具有如下技术效果或优点:
[0034]
1.本发明氢化氧杂蒽酮衍生物,提供五种新的氢化氧杂蒽酮衍生物
[0035]
phomopsisxanthones h~l,五种氢化氧杂蒽酮衍生物具有良好的抗植物病原菌和抗氧化活性,为开发农用抗生素和抗氧化药物提供新的选择。
[0036]
2.本发明氢化氧杂蒽酮衍生物的制备方法,采用化学表观遗传修饰策略,在菌株phomopsis sp.ye3350的发酵培养基中添加化学表观遗传修饰剂丁酸钠,利用化学表观遗传修饰剂丁酸钠诱导产生五种新的氢化氧杂蒽酮衍生物phomopsisxanthones h~l,该方法具有周期短、培养条件温和、副产物少、立体选择性强、成本低的优点,且具有较高的经济价值,成本低、操作简便,易于大规模生产,为获得天然来源的氢化氧杂蒽酮衍生物提供了新途径。
附图说明
[0037]
为了更清楚地说明本发明实施例中的技术方案,下面将对实施例描述中所需要使用的附图作一简单地介绍,显而易见地,下面描述中的附图是本发明的一些实施例,对于本领域普通技术人员来讲,在不付出创造性劳动的前提下,还可以根据这些附图获得其它的附图。
[0038]
图1为本发明所述化合物phomopsisxanthone h的1h
‑
nmr谱;
[0039]
图2为本发明所述化合物phomopsisxanthone h的
13
c
‑
nmr和dept谱;
[0040]
图3为本发明所述化合物phomopsisxanthone i的1h
‑
nmr谱;
[0041]
图4为本发明所述化合物phomopsisxanthone i的
13
c
‑
nmr和dept谱;
[0042]
图5为本发明所述化合物phomopsisxanthone j的1h
‑
nmr谱;
[0043]
图6为本发明所述化合物phomopsisxanthone j的
13
c
‑
nmr和dept谱;
[0044]
图7为本发明所述化合物phomopsisxanthone k的1h
‑
nmr谱;
[0045]
图8为本发明所述化合物phomopsisxanthone k的
13
c
‑
nmr和dept谱;
[0046]
图9为本发明所述化合物phomopsisxanthone l的1h
‑
nmr谱;
[0047]
图10为本发明所述化合物phomopsisxanthone l的
13
c
‑
nmr和dept谱;
[0048]
图11为本发明所述化合物phomopsisxanthone h的x
‑
单晶衍射晶体结构图;
[0049]
图12为本发明所述化合物phomopsisxanthone i的x
‑
单晶衍射晶体结构图。
[0050]
图13为本发明氢化氧杂蒽酮衍生物phomopsisxanthones h~l的结构式。
具体实施方式
[0051]
下文将结合具体实施方式和实施例,具体阐述本发明,本发明的优点和各种效果将由此更加清楚地呈现。本领域技术人员应理解,这些具体实施方式和实施例是用于说明本发明,而非限制本发明。
[0052]
在整个说明书中,除非另有特别说明,本文使用的术语应理解为如本领域中通常所使用的含义。因此,除非另有定义,本文使用的所有技术和科学术语具有与本发明所属领域技术人员的一般理解相同的含义。若存在矛盾,本说明书优先。
[0053]
除非另有特别说明,本发明中用到的各种原材料、试剂、仪器和设备等,均可通过市场购买得到或者可通过现有方法制备得到。
[0054]
本发明实施例提供的技术方案为解决上述技术问题,总体思路如下:
[0055]
近年来,随着全基因测序手段的快速发展,已发现真菌中有大量编码化合物的基因簇存在。研究表明在实验室常规培养的条件下,真菌基因组序列编码次级代谢产物的生物合成基因簇大多是沉默的。通过基因工程的技术手段使微生物中这些沉默的基因簇进行表达以产生更多的代谢产物已经成为研究天然产物的热点。目前通过修饰真菌表观基因组来探索次级代谢产物多样性是有效解决问题的途径。化学表观遗传修饰方法可以抑制影响真菌表观遗传的酶,激活沉默的生物合成基因诱导产生不同的次级代谢产物,极大地增加真菌次级代谢产物的多样性。
[0056]
基于此,本发明采用化学表观遗传修饰策略,在菌株phomopsis sp.ye3350的发酵培养基中添加化学表观遗传修饰剂丁酸钠,利用化学表观遗传修饰剂丁酸钠诱导产生五种新的氢化氧杂蒽酮衍生物phomopsisxanthones h~l,这五种氢化氧杂蒽酮衍生物具有良好的抗植物病原菌和抗氧化活性,为开发农用抗生素和抗氧化药物提供新的选择。
[0057]
下面将结合实施例及实验数据对本技术氢化氧杂蒽酮衍生物及其制备方法进行详细说明。
[0058]
实施例1
[0059]
氢化氧杂蒽酮衍生物phomopsisxanthones h~l的分离制备
[0060]
(1)配制pdb培养基:称量去皮马铃薯200g,切成小块,加入1000ml蒸馏水煮沸,纱布过滤,得马铃薯滤液,加入葡萄糖20g,ph自然,121℃灭菌30min,制得pdb培养基备用。其中pdb培养基中马铃薯的浓度为0.2g/ml,葡萄糖的浓度为0.02g/ml。
[0061]
配制改良pdb培养基:称量去皮马铃薯200g,切成小块,加入1000ml蒸馏水煮沸,纱布过滤,得马铃薯滤液,加入葡萄糖20g,磷酸氢二钾3g,硫酸镁3g,维生素b
1 10mg,ph自然121℃灭菌30min,制得改良pdb培养基备用。其中改良pdb培养基中马铃薯的浓度为0.2g/ml,葡萄糖的浓度为0.02g/ml。
[0062]
(2)将phomopsis sp.ye3350菌株接入pdb培养基中,在28
±
2℃、200r/min摇床培养4d,得到种子液。
[0063]
(3)将化学表观遗传修饰剂丁酸钠溶解于无菌水中,经孔径为0.22μm的微孔滤膜过滤得到丁酸钠溶液,其中丁酸钠溶液中丁酸钠浓度为400mm;
[0064]
(4)将步骤(3)丁酸钠溶液加入到改良pdb发酵培养基中得到含化学表观遗传修饰剂的改良pdb发酵培养基,再将种子液接入发酵培养基改良pdb培养基中,于28
±
2℃、200r/min摇床培养7~9d,得发酵产物,其中含化学表观遗传修饰剂的pdb发酵培养基中丁酸钠的浓度为500μm,种子液的接种量为10%。
[0065]
(5)将步骤(4)得到的发酵产物用纱布过滤,得发酵液和菌丝体,发酵液用乙酸乙酯萃取,菌丝体用甲醇浸提,合并提取液,减压浓缩得发酵粗提物。
[0066]
(6)将发酵粗提物用硅胶柱(200
‑
300目)进行色谱分离,用氯仿
‑
甲醇体系从体积比10:1至1:1进行梯度洗脱,收集氯仿
‑
甲醇体系体积比9:1洗脱得到的粗组分fr.1,收集氯仿
‑
甲醇体系体积比8:2洗脱得到的粗组分fr.2,收集氯仿
‑
甲醇体系体积比7:3洗脱得到的粗组分fr.3。
[0067]
粗组分fr.1用氯仿
‑
甲醇体系从体积比15:1至7:3洗脱,收集氯仿
‑
甲醇体系体积比9:1时洗脱得到的馏分,在丙酮与甲醇体积比为3:1混合溶剂中重结晶,可制备得到化合物phomopsisxanthone h。粗组分fr.2用石油醚
‑
乙酸乙酯体系从体积比9:1至7:3洗脱,收集石油醚
‑
乙酸乙酯体系体积比为9:1时洗脱得到的馏分,在甲醇与水体积比为8:2混合溶剂中重结晶,可制备得到化合物phomopsisxanthone i。粗组分fr.3用甲醇
‑
水体系从体积比1:1至9:1进行梯度洗脱,收集甲醇
‑
水体积比为6:4时洗脱得到的馏分,可制备得到化合物phomopsisxanthone j;收集甲醇
‑
水体积比为7:3时洗脱得到的馏分,可制备得到化合物phomopsisxanthone k;收集甲醇
‑
水体积比为8:2时洗脱得到的馏分,可制备得到化合物phomopsisxanthone l。
[0068]
实施例2
[0069]
氢化氧杂蒽酮化合物phomopsisxanthone h~l的结构鉴定,结果如图13所示。
[0070]
氢化氧杂蒽酮化合物phomopsisxanthone h的结构鉴定
[0071]
由实施例1制备得到的化合物phomopsisxanthone h,通过1d/2d nmr(一维核磁共振波谱和二维核磁共振波谱)以及hresi
‑
ms(高分辨电喷雾电离质谱)鉴定其结构。phomopsisxanthone h的分子式为c
15
h
16
o8;hresims m/z 309.0965[m+h]
+
,不饱和度为8。
[0072]
如图1、2,1h
‑
nmr图谱显示一个羟基质子(δ
h 11.68),一个甲基质子(δ
h 2.27),一个甲氧基质子(δ
h 4.63),两个芳香族质子(δ
h 6.30;δ
h 6.28),两个亚甲基(δ
h 3.83和3.22,δ
h 2.49和2.99),一个连氧亚甲基(δ
h 3.84),一个次甲基(δ
h 4.94)和一个连氧次甲基(δ
h 4.57)。
13
c dept
‑
nmr图谱显示15个碳信号,分别为两个羰基碳(δ
c 196.0、174.5),五个季碳(δ
c 161.5、159.2、150.4、105.4、82.4),四个次甲基(δ
c 109.3、108.9、87.8、67.2),一个连氧亚甲基(δ
c 61.8),两个亚甲基(δ
c 37.7、36.9)和一个甲基(δ
c
21.4)。1h
‑1h cosy图谱显示了两组间位芳族氢质子,表明存在两个1,2,3,5
‑
四取代的芳环。在它的hmbc图谱中观察到ch3‑
11对c
‑
1,c
‑
2,c
‑
4有相关,表明了与甲基在c
‑
3位。在hmbc光谱中,ch2‑
12与c
‑
8a(δ
c 37.7)、c
‑
10a(δ
c 82.4)和c
‑
5(δ
c 87.8)有相关点的存在,可确定了连氧亚甲基c
‑
12与b环的连接。1h
‑1h cosy光谱数据显示,连氧次甲基质子h
‑
6与亚甲基h
‑
7(δ
h 2.99)和次甲基h
‑
5(δ
h 4.94)相耦合,这表明存在γ
‑
内酯部分。从hmbc图谱中可以观察到c
‑
8(δ
c 174.5)和h
‑
5具有相关点,h
‑
5与c
‑
10a和c
‑
12有相关点,这也证实了γ
‑
内酯部分的存在,同时表明了
γ
‑
内酯部分与b环的连接。noesy图谱显示ch2oh
‑
12在c
‑
10a处是α取向,6
‑
oh在c
‑
6处是β取向。经过尝试多种溶剂系统及比例,我们最终通过丙酮:甲醇(3:1)的体系获得单晶。如图11,经过x
‑
单晶衍射确定化合物的绝对构型。经检索为新的氧杂蒽酮化合物,命名为phomopsisxanthone h。其结构式如下式(i)所示:
[0073][0074]
氢化氧杂蒽酮化合物phomopsisxanthone i的结构鉴定
[0075]
由实施例1制备得到的化合物phomopsisxanthone i,通过1d/2d nmr(一维核磁共振波谱和二维核磁共振波谱)以及hresi
‑
ms(高分辨电喷雾电离质谱)鉴定其结构。phomopsisxanthone i的分子式为c
15
h
16
o8;hresims m/z 347.0737[m+na]
+
,不饱和度为8。
[0076]
如图3、4,1h
‑
nmr图谱显示两个芳香族质子(δ
h 6.87、6.66),一个活泼的羟基质子(δ
h
12.64),一个连氧亚甲基(δ
h 3.98),四个羟基质子(δ
h 6.12、5.35、4.72、4.86)和一个甲基质子(δ
h 2.40)。
13
c dept
‑
nmr图谱显示有15个碳原子,包括一个羰基碳(δ
c 183.6),七个芳香季碳(δ
c 160.2、147.9、108.5、156.1、117.0、165.6、72.5),一个甲基碳(δ
c
22.3),一个亚甲基(δ
c 63.8)和五个次甲基(δ
c 111.9、107.6、69.4、70.0、75.0)。它与化合物phomopsisxanthone h比较相似,都有两个间位的芳香次甲基,且甲基与c
‑
3相连。结合二维图谱1h
‑1h cosy、hmqc和hmbc结果,可以推断此化合物具有与化合物phomopsisxanthone h相同的a环和b环。在hmbc图谱中可以看出5
‑
oh(δ
h 6.12)与c
‑
10a(δ
c 165.6)、c
‑
5(δ
c 69.4)、c
‑
6(δ
c 70.0)和c
‑
7(δ
c
75.0)之间的相关性,证明了5
‑
oh与c
‑
5的连接方式。根据它的noesy图谱中观察到5
‑
oh与6
‑
h相关,结合6
‑
oh与5
‑
h以及h
‑
7与h
‑
6的相关性,说明5
‑
oh与6
‑
oh呈反式构型,确定了7
‑
oh、6
‑
oh和8
‑
oh是α取向,5
‑
oh和8
‑
ch2oh是β取向。利用水:甲醇(4:1)的体系,获得化合物的单晶。如图12,经过x
‑
ray确定化合物的绝对构型,经检索为新的氧杂蒽酮类化合物,命名为phomopsisxanthone i。其结构式如下式(ii)所示:
[0077][0078]
氢化氧杂蒽酮化合物phomopsisxanthone j的结构鉴定
[0079]
由实施例1制备得到的化合物phomopsisxanthone j,通过1d/2d nmr(一维核磁共振波谱和二维核磁共振波谱)以及hresi
‑
ms(高分辨电喷雾电离质谱)鉴定其结构。
phomopsisxanthone j的分子式为c
15
h
16
o8;hresims m/z 347.0737[m+na]
+
,不饱和度为8。
[0080]
hresi
‑
ms显示分子离子峰为m/z:347.0737[m+na]
+
。如图5、6,综合nmr图谱和质谱的数据推断其分子式为c
15
h
16
o8。此化合物和化合物phomopsisxanthone i的一维1h
‑
nmr和
13
c
‑
nmr图谱数据相似,综合比较它们的一维和二维图谱,发现它们具有相同的平面结构,其具有8个不饱和度,c
‑
5、c
‑
6、c
‑
7和c
‑
8都有羟基基团的取代。从其氢谱中可以看出化合物的6
‑
oh、7
‑
oh、12α
‑
h和8
‑
oh向低场位移至δ
h 5.58、δ
h 5.26、δ
h 4.10和δ
h 5.00,12
‑
oh向高场位移至δ
h 4.77,说明化合物在c
‑
8处有不同的空间构型。noesy图谱显示h
‑
5、8
‑
oh以及7
‑
oh之间的相关点确定化合物的8
‑
ch2oh取向与phomopsisxanthone i的取向不同,为α取向。经检索为新化合物,命名为phomopsisxanthone j。其结构式如下式(iii)所示:
[0081][0082]
氢化氧杂蒽酮化合物phomopsisxanthone k的结构鉴定
[0083]
由实施例1制备得到的化合物phomopsisxanthone k,通过1d/2d nmr(一维核磁共振波谱和二维核磁共振波谱)以及hresi
‑
ms(高分辨电喷雾电离质谱)鉴定其结构。phomopsisxanthone k的分子式为c
15
h
14
o7;hresims m/z 305.0601[m
‑
h]
‑
,不饱和度为9。
[0084]
如图7、8,1h
‑
nmr图谱数据在δ
h 11.90显示一个活泼的羟基质子,此外还显示一个甲基质子(δ
h 2.41),一个亚甲基质子(δ
h 3.72,3.66),5个次甲基质子(δ
h 6.97,6.72,4.28,4.26,4.14),还有3个羟基质子(δ
h 6.60,6.03,5.40)。图谱显示15个碳原子,1个羰基碳(δ
c 184.2),7个芳香季碳(δ
c 165.9,159.6,156.4,148.8,115.6,108.4,77.9),一个甲基(δ
c 22.3),一个亚甲基(δ
c 75.2),5个次甲基(δ
c 74.3,72.6,70.2,112.5,108.2)。通过比较它与化合物phomopsisxanthone j的一维图谱发现它们比较相似,都有两个间位的芳香次甲基,且甲基与c
‑
3相连。结合1h
‑1h cosy、hmqc和hmbc图谱结果表明化合物phomopsisxanthone k具有与化合物phomopsisxanthone j相同的a环和b环。分析a环和b环的结构以及不饱和度,表明化合物除了两个苯环和一个双键外,还应该有两个环。在δ
h 6.60和5.40处都显示双重峰,表明羟基质子5
‑
oh和7
‑
oh与次甲基相连。从hmbc图谱可以看出5
‑
oh与c
‑
5和c
‑
10a有相关点,7
‑
oh与c
‑
7和c
‑
8有相关点,由此推断5
‑
oh和7
‑
oh与环c的连接方式。noesy图谱没有显示h2‑
12与7
‑
oh和5
‑
oh存在相关点,7
‑
oh和5
‑
oh也没有相关点存在,8
‑
oh与ch3‑
11有相关点,推断出5
‑
oh为α取向,7
‑
oh为β取向。经检索为新化合物,命名为phomopsisxanthone k。其结构式如下式(iv)所示:
[0085][0086]
氢化氧杂蒽酮化合物phomopsisxanthone l的结构鉴定
[0087]
由实施例1制备得到的化合物phomopsisxanthone l,通过1d/2d nmr(一维核磁共振波谱和二维核磁共振波谱)以及hresi
‑
ms(高分辨电喷雾电离质谱)鉴定其结构。phomopsisxanthone l的分子式为c
15
h
14
o6;hresims m/z 313.0683[m+na]
+
‑
,不饱和度为9。
[0088]
如图9、10,1h
‑
nmr图谱显示了一个甲基质子(δ
h 2.38),一个亚甲基(δ
h 4.03,3.60),六个次甲基(δ
h 3.66,4.20,4.25,4.63,6.53,6.75)。
13
c dept
‑
nmr图谱显示15个碳原子,包括一个羰基碳(δ
c 181.5),6个芳香季碳(δ
c 159.9,147.4,163.8,156.7,115.9,108.1),一个甲基(δ
c 21.0),一个亚甲基(δ
c 71.5),六个次甲基(δ
c 35.0,69.6,69.9,74.2,111.2,107.3)。通过对比phomopsisxanthone k与化合物phomopsisxanthone l的图谱,发现它们相似,其主要区别是化合物phomopsisxanthone l在δ
h 3.66处多了一个峰,而h
‑
7向低场位移至δ
h 4.63处。分子量比化合物phomopsisxanthone k少了16,可推测h
‑
7邻位的羟基取代变成了氢取代。从hmqc图谱中可以看出h
‑
8与c
‑
8a、c
‑
6和c
‑
8存在相关点,1h
‑1h cosy图谱显示h
‑
8与h
‑
12α和h
‑
7有相关点,表明了c
‑
8与c
‑
12和c
‑
7的连接方式。从noesy图谱中可以观察到h
‑
7与h
‑
8和h
‑
6有相关点的存在,h
‑
8与h
‑
12α有相关点的存在,h
‑
5与h
‑
7没有相关点,表明h
‑
8、h
‑
6以及5
‑
oh是α取向,7
‑
oh是β取向。经检索为新化合物,命名为phomopsisxanthone l。其结构式如下式(v)所示:
[0089][0090]
本发明五个氢化氧杂蒽酮衍生物均溶解于甲醇、吡啶、二甲亚砜等,不溶于石油醚和水。化合物phomopsisxanthones h~l的1h nmr和
13
c nmr数据见表1。
[0091]
表1氢化氧杂蒽酮衍生物phomopsisxanthones h~l的1h nmr和
13
c nmr数据
[0092][0093][0094]
phomopsisxanthone h的测定溶剂为氘代丙酮;phomopsisxanthone i、j、l的测定溶剂为氘代二甲亚砜;phomopsisxanthone k的测定溶剂为氘代甲醇。
[0095]
实施例3
[0096]
氢化氧杂蒽酮化合物phomopsisxanthones h~l的抗植物病原菌活性和抗氧化能力测定:
[0097]
(1)抗植物病原菌的活性测定:
[0098]
4种植物病原指示菌包括番茄灰霉菌(botrytis cinerea)、小麦赤霉菌(gibberella saubinetii)、马铃薯腐皮镰刀菌(fusarium solani)和新月弯孢霉(curvularia lunata)。
[0099]
在无菌环境下,将生长良好的病原菌接入每瓶含有100ml液体培养基的250ml锥形瓶中。称取1024μg化合物后用dmso溶液溶解,在96孔板上用无菌水倍比稀释成516
‑
2μg/ml。每孔加入50μl菌悬液,吸打混合均匀。阳性对照为制霉菌素,每组病原菌做2组平行实验。在28℃培养24
‑
48h,每8h观察结果并记录一次。
[0100]
(2)dpph自由基清除能力:
[0101]
称1mg待测样品,加入2ml dmso溶解得到0.5mg/ml样品母液。然后再将所配制的母液稀释成5
‑
50μg/ml。称取7.886mg的dpph(1,1
‑
二苯基
‑2‑
三硝基苯肼)用100ml dmso溶液进行溶解,得到浓度为0.2mm的dpph母液。吸取1ml配置好的dpph母液加入不同浓度(3ml,5
‑
50μg/ml)的化合物溶液中,在暗条件下反应30分钟。取反应后的样品200μl于96孔板中,用酶标仪测定其在517nm处的吸收值。以dmso为阴性对照,维生素c(v
c
)和维生素e(v
e
)为阳性对照。
[0102]
将所测得数据用公式[(ac
–
as)/ac]
×
100%分别计算出化合物的dpph自由基清除率k(%)。ac为阴性对照dmso在517nm处测定的吸收值,as为加入化合物或者阳性对照v
c
和v
e
在517nm处测定的吸收值。根据其自由清除率数据计算出该化合物的ic
50
(半数抑制浓度)值。
[0103]
(3)abts
+
自由基清除能力:
[0104]
称取1mg待测样品,加入2ml dmso溶解得到0.5mg/ml样品母液。然后再将所配制的母液稀释成5
‑
50μg/ml。称取109.736mg abts
+
(2,2
‑
联氮
‑
二(3
‑
乙基
‑
苯并噻唑
‑6‑
磺酸)二铵盐)和66.2284mg的过硫酸钾用100ml水进行溶解,在室温下避光反应12小时后得到abts
+
浓度为0.2mm的母液。吸取1ml配置好的abts
+
母液加入稀释好的不同浓度(3ml,5
‑
50g/ml)化合物溶液中,在暗条件下反应10分钟。取200μl反应后的样品于96孔板中,用酶标仪测定其在734nm处的吸收值。在本试验中dmso为阴性对照,v
c
和v
e
为阳性对照。将所测得数据用公式[(ac
–
as)/ac]
×
100%分别计算出化合物的dpph自由基清除率k(%)。ac为阴性对照dmso在517nm处测定的吸收值,as为加入化合物或者阳性对照v
c
和v
e
在734nm处测定的吸收值。根据其自由清除率数据计算出该化合物的ic
50
值。
[0105]
本发明所述的氢化氧杂蒽酮衍生物phomopsisxanthones h~l对4种病原指示菌的最低抑菌浓度(mic)值如表2所示。
[0106]
表2化合物phomopsisxanthones h~l对4种病原指示菌的抑制活性
[0107][0108]
本发明所述的氢化氧杂蒽酮衍生物phomopsisxanthones h~l对dpph自由基和abts
+
自由基清除作用的半数抑制浓度(ic
50
)值如表3所示。
[0109]
表3化合物phomopsisxanthones h~l的自由基清除能力
[0110][0111]
实验结果表明,本发明所述的氢化氧杂蒽酮化合物phomopsisxanthones h~l对4种植物病原指示菌都具有不同程度的抗菌活性,尤其是化合物phomopsisxanthone h的抗菌活性最强,化合物phomopsisxanthone i和phomopsisxanthone j对小麦赤霉菌也显示较强的活性。所测化合物对dpph自由基和abts
+
自由基都具有清除能力,其中化合物phomopsisxanthone k和phomopsisxanthone l显示最强的清除能力,表现出较强的抗氧化活性。因此,本发明所述的氢化氧杂蒽酮化合物phomopsisxanthones h~l具有作为制备新的农用抗生素和抗氧化药物的潜在用途。
[0112]
本发明实施例中的一个或多个技术方案,至少具有如下技术效果或优点:
[0113]
(1)本发明氢化氧杂蒽酮衍生物的制备方法,在制备发酵粗提物过程中,发酵液用乙酸乙酯萃取,菌丝体用甲醇浸提,其目的在于提取获得发酵液和菌丝体中的有效组分,带来的好处是将发酵产物中的有效物质与未利用完的培养基成分和水溶性杂质分开。
[0114]
(2)本发明氢化氧杂蒽酮衍生物的制备方法,拟茎点霉菌的发酵培养基采用改良后的pdb培养基,相比现有常规pdb培养基而言,其带来的有益效果是能产生与常规pdb培养基不同的发酵产物,原理在于培养基成分的改变会影响微生物发酵产物的合成。
[0115]
(3)本发明氢化氧杂蒽酮衍生物的制备方法,拟茎点霉菌的发酵培养基中,丁酸钠浓度为200~500μm的好处在于该浓度范围能够诱导本发明化合物的产生。
[0116]
(4)本发明氢化氧杂蒽酮衍生物的制备方法,在收集粗组分fr.1
‑
fr.3,以及分离phomopsisxanthones h~l过程中,选择的各洗脱剂体系及洗脱剂的不同体积比,目的在于获得对应的目标馏分或目标产物,若采用其他的洗脱剂体系或其他的体积比,则获得目标产物phomopsisxanthones h~l的分离流程将会比现有技术繁琐,耗时更长或难以获得。
[0117]
最后,还需要说明的是,术语“包括”、“包含”或者其任何其他变体意在涵盖非排他性的包含,从而使得包括一系列要素的过程、方法、物品或者设备不仅包括那些要素,而且还包括没有明确列出的其他要素,或者是还包括为这种过程、方法、物品或者设备所固有的要素。
[0118]
尽管已描述了本发明的优选实施例,但本领域内的技术人员一旦得知了基本创造性概念,则可对这些实施例作出另外的变更和修改。所以,所附权利要求意欲解释为包括优
选实施例以及落入本发明范围的所有变更和修改。
[0119]
显然,本领域的技术人员可以对本发明进行各种改动和变型而不脱离本发明的精神和范围。这样,倘若本发明的这些修改和变型属于本发明权利要求及其等同技术的范围之内,则本发明也意图包含这些改动和变型在内。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1