用于多核苷酸测序的组合物和方法与流程
用于多核苷酸测序的组合物和方法
1.本技术是申请日为2014年11月26日,申请号为201480064725.5,发明名称为“用于多核苷酸测序的组合物和方法”的申请的分案申请。
2.相关申请的引用
3.本专利申请主张2013年11月26日提交的标题为“用于多核苷酸测序的组合物和方法”的美国临时专利申请no.61/909,316的权益,该专利申请的全部内容作为参考并入本文。
4.序列表
5.本专利申请包含以ascii格式电子提交的序列表,并且该序列表以其全部内容作为参考并入本文。创建于2014年11月25日的所述ascii拷贝的名称为12957
‑
139
‑
228_sl.txt和大小为19,778字节。
6.发明背景
7.本发明公开一般地涉及表征目标多核苷酸的方法和组合物,其包括表征目标多核苷酸的序列。
8.由于多核苷酸(例如,dna或rna)中编码的信息对医学和生命科学非常重要,因此需要快速且经济地对多核苷酸测序。目前,商品化测序技术需要样品和文库制备,两者均很费力。此外,对于多种应用,读取数据比预期更慢。因此,通量受限并且成本相对较高。纳米孔测序代表了正在开发的用于快速且廉价地对目标多核苷酸测序的一种新方法。
9.纳米孔测序利用了可以为离子电流提供通道的纳米孔。电泳驱动多核苷酸通过纳米孔,并且由于多核苷酸穿过纳米孔,因此它降低了通过纳米孔的电流。每个通过的核苷酸或一系列核苷酸获得了特征电流,并且对电流电平的记录对应于多核苷酸序列。由于一些电流电平取决于多个核苷酸(通常3
‑
4个),因此仍需要改善目前工艺水平以改善准确度。由于多核苷酸易位通过纳米孔所获得的电流电平的任何其它信息可以提供优势,如形状和持久时间。
10.纳米孔测序的常见问题是多核苷酸通过纳米孔的易位过快以至于单个核苷酸的电流电平(current level)过短而难以分辨。纳米孔测序的一种方法涉及在抗电压电位的多核苷酸结合蛋白的引导下控制多核苷酸通过纳米孔的易位,所述结合蛋白如解旋酶、易位酶或聚合酶。尽管有这种控制易位,但是一些测序错误形式仍存在并且导致较差的测序准确度。
11.因此,仍需要提供进一步控制多核苷酸通过纳米孔的易位并且在核苷酸差异中更好地分辨核苷酸易位的方法和组合物。本发明公开满足了该需要并提供了相关优势。
12.发明概述
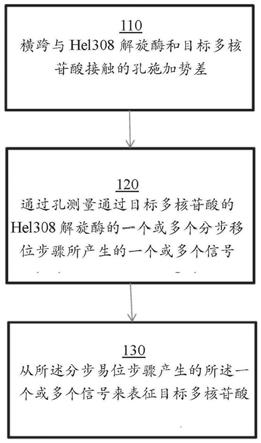
13.提供了表征目标多核苷酸的方法。所述方法包括:(a)跨越与hel308解旋酶(helicase)和目标多核苷酸接触的孔施加势差;(b)测量目标多核苷酸通过孔的一个或多个分步易位步骤产生的一个或多个信号;和(c)根据分步易位步骤的电信号,表征目标多核苷酸。目标多核苷酸的表征包括鉴别以下中的一个或多个:(1)目标多核苷酸的序列;(2)目标多核苷酸的修饰;(3)目标多核苷酸的长度;(4)目标多核苷酸的身份;(5)目标多核苷酸
id no:1具有至少15%、至少20%、至少25%、至少30%、至少35%、至少40%、至少45%、至少50%、至少55%、至少60%、至少65%或至少70%同源性的氨基酸序列。
37.项目20.根据项目1所述的方法,其中,所述孔为固态孔。
38.项目21.根据项目1所述的方法,其中,所述孔为生物和固态杂交孔。
39.项目22.根据项目21所述的方法,其中,所述生物和固态杂交孔为多肽
‑
固态杂交孔。
40.项目23.根据项目21所述的方法,其中,所述生物和固态杂交孔为多核苷酸
‑
固态杂交孔。
41.项目24.根据项目1所述的方法,其中,所述hel308解旋酶为表1和2所示的解旋酶或其变体。
42.项目25.根据项目1所述的方法,其中,所述目标多核苷酸选自单链多核苷酸、双链多核苷酸和部分双链多核苷酸。
43.项目26.调节目标多核苷酸通过孔的分步易位步骤的方法,所述方法包括:
44.(a)跨越与hel308解旋酶和目标多核苷酸接触的孔施加势差;
45.(b)将所述hel308解旋酶与一定浓度的hel308解旋酶底物接触,所述hel308解旋酶底物的浓度不同于所述底物的参比浓度,所述底物浓度在分步易位步骤持续时间中所产生的变化与所述底物浓度与所述参比浓度相比的差异成正比,和
46.(c)测量通过所述hel308解旋酶的所述目标多核苷酸通过所述孔的一个或多个分步易位步骤所产生的一个或多个信号。
47.项目27.根据项目26所述的方法,还包括根据通过所述一个或多个分步易位步骤产生的一个或多个信号,表征所述目标多核苷酸。
48.项目28.根据项目27所述的方法,其中,表征所述目标多核苷酸包括鉴别以下中的一种或多种:所述目标多核苷酸的序列、所述目标多核苷酸的修饰、所述目标多核苷酸的长度、所述目标多核苷酸的身份、所述目标多核苷酸的来源和所述目标多核苷酸的二级结构。
49.项目29.根据项目26所述的方法,其中,所述势差包括电势差。
50.项目30.根据项目26所述的方法,其中,所述一个或多个信号包括电信号。
51.项目31.根据项目26所述的方法,其中,所述一个或多个信号包括光信号。
52.项目32.根据项目26所述的方法,其中,所述底物浓度为所述hel308解旋酶底物的半饱和浓度。
53.项目33.根据项目26所述的方法,其中,所述参比浓度为所述hel308解旋酶底物的饱和浓度。
54.项目34.根据项目26所述的方法,其中,所述底物和所述参比浓度两者均不是所述底物的饱和浓度。
55.项目35.根据项目26所述的方法,其中,所述底物浓度和所述参比浓度是所述hel308解旋酶底物的半饱和浓度。
56.项目36.根据项目26所述的方法,其中,所述hel308解旋酶底物为三磷腺苷(atp)。
57.项目37.根据项目26所述的方法,其中,所述分步易位步骤包括所述hel308解旋酶的完整易位循环的第一分步易位步骤。
58.项目38.根据项目26所述的方法,其中,所述分步易位步骤包括所述hel308解旋酶
的完整易位循环的第二分步易位步骤。
59.项目39.根据项目26所述的方法,其中,所述目标多核苷酸的易位处于通过势差施加于易位通过所述孔的多核苷酸上的外加作用力的相反方向。
60.项目40.根据项目26所述的方法,其中,所述目标多核苷酸的易位处于通过势差施加于易位通过所述孔的多核苷酸上的外加作用力的方向。
61.项目41.根据项目30所述的方法,其中,所述电信号为选自电流、电压、隧穿、电阻、电位、电压、电导率和横向电测量的测量。
62.项目42.根据项目41所述的方法,其中,所述电信号包括穿过所述孔的电流。
63.项目43.根据项目30所述的方法,其中,使用得自完整易位循环的两个分步步骤的电信号表征所述目标多核苷酸中的一个或多个核苷酸残基,与使用得自完整易位循环的单个电信号的一个或多个核苷酸的表征相比,其准确度大于50%。
64.项目44.根据项目26所述的方法,其中,与所述参比浓度相比,在较低底物浓度时,表征所述目标多核苷酸中的一个或多个核苷酸残基具有更大的准确度。
65.项目45.根据项目26所述的方法,其中,所述孔为生物孔。
66.项目46.根据项目45所述的方法,其中,所述生物孔为多肽孔。
67.项目47.根据项目45所述的方法,其中,所述生物孔为多核苷酸孔。
68.项目48.根据项目46所述的方法,其中,所述多肽孔具有5个核苷酸或以下的缢缩区。
69.项目49.根据项目46所述的方法,其中,所述多肽孔包含耻垢分枝杆菌孔蛋白a(mspa)。
70.项目50.根据项目49所述的方法,其中,所述mspa具有seq id no:1所示或与seq id no:1具有至少15%、至少20%、至少25%、至少30%、至少35%、至少40%、至少45%、至少50%、至少55%、至少60%、至少65%、至少70%、至少75%、至少80%、至少85%、至少90%、至少95%或至少99%同源性的氨基酸序列。
71.项目51.根据项目26所述的方法,其中,所述孔为固态孔。
72.项目52.根据项目26所述的方法,其中,所述孔为生物和固态杂交孔。
73.项目53.根据项目52所述的方法,其中,所述生物和固态杂交孔为多肽
‑
固态杂交孔。
74.项目54.根据项目52所述的方法,其中,所述生物和固态杂交孔为多核苷酸
‑
固态杂交孔。
75.项目55.根据项目26所述的方法,其中,所述hel308解旋酶为表1和2所示的解旋酶或其变体。
76.项目56.根据项目26所述的方法,其中,所述目标多核苷酸选自单链多核苷酸、双链多核苷酸和部分双链多核苷酸。
77.项目57.表征目标多核苷酸的组合物,其包含孔、hel308解旋酶和包含在含有小于1mm atp或含有核苷酸类似物的溶液中的目标多核苷酸。
78.项目58.根据项目57所述的组合物,其中,含有小于1mm atp的所述溶液包括以下浓度,其选自:0.1μm、1.0μm、10μm、100μm、0.5mm和0.9mm atp。
79.项目59.根据项目57所述的组合物,其中,所述孔为生物孔。
80.项目60.根据项目59所述的组合物,其中,所述生物孔为多肽孔。
81.项目61.根据项目59所述的组合物,其中,所述生物孔为多核苷酸孔。
82.项目62.根据项目60所述的组合物,其中,所述多肽孔具有5个核苷酸或以下的缢缩区。
83.项目63.根据项目58所述的组合物,其中,所述多肽孔包含耻垢分枝杆菌孔蛋白a(mspa)。
84.项目64.根据项目63所述的组合物,其中所述mspa具有seq id no:1所示或与seq id no:1具有至少15%、至少20%、至少25%、至少30%、至少35%、至少40%、至少45%、至少50%、至少55%、至少60%、至少65%、至少70%、至少75%、至少80%、至少85%、至少90%、至少95%或至少99%同源性的氨基酸序列。
85.项目65.根据项目57所述的组合物,其中,所述孔为固态孔。
86.项目66.根据项目57所述的组合物,其中,所述孔为生物和固态杂交孔。
87.项目67.根据项目66所述的组合物,其中,所述生物和固态杂交孔为多肽
‑
固态杂交孔。
88.项目68.根据项目66所述的组合物,其中,所述生物和固态杂交孔为多核苷酸
‑
固态杂交孔。
89.项目69.根据项目57所述的组合物,其中,所述hel308解旋酶为表1和表2所示的解旋酶或其变体。
90.项目70.根据项目57所述的组合物,其中,所述目标多核苷酸选自单链多核苷酸、双链多核苷酸和部分双链多核苷酸。
91.项目71.根据项目1所述的方法,其中,所述表征包括应用改进型维特比算法。
92.项目72.根据项目1所述的方法,还包括:
93.(d)在步骤(c)后,改变至少一种参数从而改变所述目标多核苷酸通过所述孔的由所述hel308解旋酶造成的一个或多个分步易位步骤的时机;和
94.(e)使用所述改变的至少一种参数,重复步骤(a)
‑
(c)。
95.项目73.根据项目72所述的方法,还包括合并步骤(c)和(e)期间产生的信号并基于所述合并的信号表征所述目标多核苷酸。
96.项目74.根据项目73所述的方法,其中,所述改变的至少一种参数选自温度、盐浓度、辅因子浓度、atp产物浓度、ph和所使用的具体的hel308解旋酶。
97.项目75.根据项目1所述的方法,其中,所述表征包括检测和识别所述一种或多种信号中的电平以及基于所述检测和鉴别的电平确定和输出所述目标多核苷酸的序列。
98.项目76.根据项目75所述的方法,其中,所述检测和识别所述一种或多种信号中的电平包括输出全电平、分电平、所有电平和电平标识符中的一个或多个。
99.项目77.根据项目76所述的方法,其中,基于所述检测和识别的电平,所述确定和输出所述目标多核苷酸的序列包括将全电平、分电平、所有电平和电平标识符中的一个或多个作为输入,基于所述输入调用多个序列,和基于所调用的多个序列的相关置信信息选择并输出至少一个所调用的序列。
100.项目78.根据项目76所述的方法,其中,基于所述检测和识别的电平,所述确定和输出所述目标多核苷酸的序列包括将全电平、分电平、所有电平和电平标识符中的一个或
多个作为输入,基于所述输入调用多个序列,和基于所调用的多个序列的部分的相关置信信息选择多个所调用序列的部分并将其彼此连接。
101.项目79.根据项目76所述的方法,其中,基于所述检测和识别的电平,所述确定和输出所述目标多核苷酸的序列包括将全电平、分电平、所有电平和电平标识符中的一个或多个作为输入,基于所述输入调用多个序列,将所调用的序列与模型序列比较,和基于所调用的序列与模型序列比较的相关置信信息选择和输出至少一个调用序列。
102.项目80.根据项目76所述的方法,其中,基于所述检测和鉴别的电平,所述确定和输出所述目标多核苷酸的序列包括将全电平、分电平、所有电平和电平标识符中的一个或多个作为输入,基于所述输入调用多个序列,将所调用的序列与模型序列比较,和基于多个调用序列的部分与模型序列比较的相关置信信息选择多个调用序列的部分并将其彼此连接。
附图说明
103.图1a示出了通过解旋酶的多核苷酸易位的静电尺蠖模型(electrostatic inchworm model)。
104.图1b示意性示出了根据一些实施方式的第一示例性组合物,其包含与hel308解旋酶接触的孔。
105.图1c示意性示出了根据一些实施方式的用于表征目标多核苷酸的示例性方法中的步骤。
106.图2a示出了根据一些实施方式的phi29聚合酶和hel308 tga解旋酶易位事件的比较。与使用phi29 dna聚合酶所观察的易位步骤相比,示出了使用hel308 tga解旋酶所观察的分步易位步骤。
107.图2b示出了根据一些实施方式的phi29聚合酶和hel308 tga解旋酶易位事件的比较。将使用phi29聚合酶作为分子马达易位通过mspa
‑
m2纳米孔的单链多核苷酸模板所产生的预期电流电平与使用hel308 tga解旋酶作为分子马达所观察到的那些相比,示出了用hel308 tga解旋酶观察到的分步易位步骤。
108.图2c示出了根据一些实施方式的phi29聚合酶和hel308 tga解旋酶易位事件的比较。对于简单重复的核苷酸序列(seq id no:74),与使用phi29 dna聚合酶所观察的易位步骤相比,示出了使用hel308 tga解旋酶所观察的分步易位步骤。
109.图3示出了根据一些实施方式对分步易位步骤所提议的“基于紧握”的机制。
110.图4a和图4b示出了根据一些实施方式atp浓度对分步易位步骤停留时间的示例性影响。
111.图5根据一些实施方式,对完整步骤(菱形)和1/2步骤(正方形)绘制了电流迹线电子产生的序列重构准确度(隐马尔可夫模型(hmm))对不同电平的附加噪声的图。
112.图6a示出了根据一些实施方式hmm解码纳米孔中序列所需的具有非零概率的状态转移,其中多核苷酸是通过马达酶移动的。所述马达为在1个核苷酸的步骤中移动多核苷酸的phi29 dnap或类似的酶。
113.图6b示出了根据一些实施方式hmm解码纳米孔中序列所需的具有非零概率的状态转移,其中多核苷酸是通过马达酶移动的。所述马达为使聚合物能够分步移动的hel308解
旋酶或类似的酶。
114.图7示出了根据一些实施方式发现电流形状作为高斯偏移的函数的预期准确度。菱形示出了具有完整核苷酸步骤的马达。圆形示出了具有分步易位步骤的马达,而正方形示出了具有与持久时间值合并的分步易位步骤的马达。
115.图8示出了根据一些实施方式随焦磷酸盐浓度的改变,hel308解旋酶活性的示例性调节。
116.图9示出了根据一些实施方式用核苷酸抑制剂原钒酸钠和用核苷酸类似物腺苷5'
‑
(β,γ
‑
亚氨基)三磷酸锂盐水合物对hel308解旋酶活性的示例性调节。
117.图10示出了根据一些实施方式使用可以通过电平(level)和电平持续时间(level duration)从两个独立序列读取获得的额外分步易位步骤所提供的信息的方法的实例。
118.图11示出了根据一些实施方式使用可以通过电平和电平持续时间从两个并行序列读取获得的额外分步易位步骤所提供的信息的方法的实例。
119.图12示出了根据一些实施方式使用通过具有或不具有持续时间信息的电流迹线的由额外分步易位步骤所提供的信息的方法的实例。
120.图13a
‑
图13e示出了根据一些实施方式基于具有hel308解旋酶3'突出结合位点和胆固醇双层锚点的三元多核苷酸复合物,通过hel308解旋酶的控制的多核苷酸易位。实心圆形(
●
)表示5'磷酸盐。实心菱形(
◆
)表示3'胆固醇。具有凹口的实心半透明圆表示hel308解旋酶。虚线表示任意长度。大的灰色箭头表示多核苷酸进入或移出孔的多核苷酸移动的方向(通过或抵抗外加场)。大的黑色箭头表示解旋酶沿多核苷酸的易位方向,其为3'至5’。孔(漏斗状圆锥形物体)位于膜(双水平线)中。
121.图14a
‑
图14d示出了根据一些实施方式基于具有hel308解旋酶3'突出结合位点和胆固醇双层锚点的三元多核苷酸复合物,通过hel308解旋酶的控制的多核苷酸易位。实心圆环(
●
)表示5'磷酸盐。实心菱形(
◆
)表示3'胆固醇。具有凹口的实心半透明圆表示hel308解旋酶。虚线表示任意长度。大的灰色箭头表示多核苷酸进入或移出孔的多核苷酸移动的方向(通过或抵抗外加场)。大的黑色箭头表示解旋酶沿多核苷酸的易位方向,其为3'至5’。孔(漏斗状圆锥形物体)位于膜(双水平线)中。符号与图13a
‑
13e中相同。在该图中,存在在多核苷酸“ii”上产生用于hel308解旋酶结合的3'突出的单个杂交多核苷酸“i”,以及含有任选的胆固醇部分。
122.图15a
‑
图15c示出了根据一些实施方式的沿梯度力的相同方向的控制的易位。具有凹口的实心半透明圆表示hel308解旋酶。虚线表示任意长度。大的灰色箭头表示通过外加场多核苷酸移动进入孔的方向。大的黑色箭头表示解旋酶沿多核苷酸的易位方向,其为3'至5’。孔(漏斗状圆锥形物体)位于膜(双水平线)中。
123.图16示意性示出了已在hel308作为其成员的sf2家族中鉴别的多个基序(按照出现顺序分别为seq id no:75
‑
81),例如,dead
‑
箱(seq id no:2)解旋酶(根据tuteja等人,“unraveling dna helicases:motif,structure,mechanism and function,”european journal of biochemistry 271(10):1849
‑
1863(2004)改编)。
124.图17a
‑
图17d示出了根据一些实施方式使用某些参数通过hel308 mbu解旋酶、hel308 tga解旋酶和phi29聚合酶易位事件产生的示例性信号。
125.图18示意性示出了根据一些实施方式实施使用表征多核苷酸条型码的分步易位
的测定的示例性方法中的步骤。
126.图19a示意性示出了用于表征来自多核苷酸通过孔的单步易位的信号的示例性隐马尔可夫模型(hmm)的方面。
127.图19b示意性示出了根据一些实施方式用于表征来自使用hel308解旋酶的多核苷酸通过孔的分步易位的信号的示例性hmm的方面。
128.图20a示出了根据一些实施方式使用分步步骤的从头序列的示例性结果。
129.图20b
‑
图20c示出了根据一些实施方式使用分步步骤的图形匹配的示例性结果。
130.图21a
‑
21c示意性示出了根据一些实施方式对于多核苷酸通过孔的不同易位,可以作为时间函数产生的信号。
131.图22a
‑
图22d示出了根据一些实施方式使用多核苷酸通过孔的分布易位所提供的信息的说明性方法中的步骤。
132.图23示出了根据一些实施方式对于适合用作各自条型码的第一说明性多核苷酸序列(seq id no:89)和第二说明性多核苷酸序列(seq id no:90),可以作为时间函数产生的示例性模拟信号。
133.图24a
‑
图24d示出了根据一些实施方式对于适合用作各自条型码的第一和第二说明性多核苷酸序列可以作为时间函数产生的示例性模拟信号。
134.图25a和图25b分别示出了根据一些实施方式对于适合用作各自条型码的第一和第二说明性多核苷酸序列可以作为时间函数产生的示例性模拟信号。
135.图26a
‑
图26d分别示出了根据一些实施方式对于适合用作各自条型码的第一和第二说明性多核苷酸序列作为时间函数产生的示例性测量信号。
136.发明详述
137.本发明公开提供了使用目标多核苷酸通过孔易位的一个或多个分步易位步骤来表征目标多核苷酸,包括表征目标多核苷酸的序列的方法和组合物。
138.在开发纳米孔测序技术时,可以在分子马达,如解旋酶、易位酶或抗势差(例如,抗由势差产生的作用力)的聚合酶的引导下,实现一定水平的多核苷酸通过纳米孔的控制易位。分子马达可以以分步方式移动多核苷酸,通常每步使用一个或多个核苷酸。这种控制的棘轮将多核苷酸通过纳米孔的易位从μsec/核苷酸的天然速率减缓至msec/核苷酸。
139.分子马达可以使用核苷酸水解的能量来驱动多核苷酸通过纳米孔的易位。解旋酶是其中atp水解是用于多核苷酸易位的能源的实例。图1中的卡通画示出了通过解旋酶的多核苷酸易位的静电尺蠖模型(electrostatic inchworm model)(参见frick等人,current pharmaceutical design,12:1315
‑
1338(2006))。在该模型中,单链多核苷酸保持在带负电荷的槽(cleft)中,其将解旋酶的两个reca结构域与第三个结构域分开。在不存在atp的情况下,书夹残基(bookend residue)(例如,hcv解旋酶中的trp501)和夹钳残基(例如,hcv解旋酶中的arg393)防止单链多核苷酸滑过槽。一旦atp结合,reca结构域旋转,从而移动带正电荷的arg
‑
夹钳。arg
‑
夹钳吸引带负电荷的单链多核苷酸,其反过来清除书夹。然后,带负电荷的槽排斥单链多核苷酸,并且单链多核苷酸易位通过解旋酶直至atp被水解。因此,在该示例性模型中,多核苷酸易位通过解旋酶包括至少两个步骤:其中解旋酶结合至atp并经历构象变化的第一步骤,和atp被水解并且多核苷酸易位通过解旋酶的第二步骤。
140.图1b示意性示出了根据一些实施方式的第一示例性组合物,其包含与hel308解旋
酶接触的孔。在图1b中,具有凹口的实心半透明圆表示如本文所提供的hel308解旋酶。直线表示多核苷酸,而虚线表示任意长度的多核苷酸。大的灰色箭头表示多核苷酸移动进入或移出孔的方向,而大的黑色箭头表示解旋酶沿多核苷酸易位的方向,其为3'至5'。在所示实施方式中,孔(漏斗状圆锥形物体)位于膜(双水平线)中,尽管可以适合地使用其它孔构型。在图1b所示的实施方式中,多核苷酸运动的方向可以与孔两侧通过势差(说明性地,180v的势差,尽管可以适合地使用其它势差)所产生的外加场相同。为了使多核苷酸移动方向与孔两侧通过势差所产生的外加场相反,因此可以翻转dna取向,如以下参考图15a
‑
15c更详细地描述的。如本文更详细地提供的,hel308解旋酶可以导致多核苷酸通过孔的分步易位,其可以有利于核苷酸的表征。例如,基于可以表征的多核苷酸,这种分步易位可以生产一个或多个信号。所述一种或多种信号可以包括如在本文其它处所描述的电信号,或者可以包括如在本文其它处所描述的光信号。示例性电信号可以选自电流、电压、隧穿、电阻、电位、电压、电导率和横向电测量的测量。
141.说明性地,由于hel308解旋酶使多核苷酸分步易位通过孔,因此孔内不同核苷酸碱基的通过可以导致通过孔的电流的可测量的变化;这种电流可以被称为“阻塞”电流。如本文更详细地描述的,可以基于信号变化,例如,基于通过孔的电流变化(这些变化基于通过hel308解旋酶的多核苷酸通过孔的分步易位步骤)确定多核苷酸的一个或多个特征,如多核苷酸序列、多核苷酸修饰、多核苷酸长度、多核苷酸身份、多核苷酸来源或多核苷酸二级结构或其任何适合的组合。在其中孔不对称(例如,包括孔口直径大于孔底)的实施方式中,hel308解旋酶可以与孔口接触,如图1b中所示。这种构型可以被称为“正向”构型。更一般地说,“正向构型”可以表示不考虑孔是否包括比孔底更宽的孔口,其中分子可以天然地通过孔的方向。作为另外一种选择,可以任意定义“正向方向”。
142.图1c示意性示出了根据一些实施方式的用于表征目标多核苷酸的示例性方法中的步骤。所述方法可以包括在与hel308解旋酶和目标多核苷酸接触的孔两侧施加势差的步骤(步骤110)。以与以下参考图13a
‑
13e和14a
‑
14d进一步描述的类似的方式,多核苷酸的易位可以处于施加在易位通过孔的多核苷酸上的由势差所引起的外加力相反的方向,或者多核苷酸的易位可以处于施加在易位通过孔的多核苷酸上的由势差所引起的外加力的方向。任选地,步骤110
‑
130可以重复一次或多次。分步易位步骤(步骤120)可以包括hel308解旋酶完整易位循环的第一分步易位步骤或可以包括hel308解旋酶完整易位循环的第二易位步骤。
143.如本文所使用的,术语“多核苷酸”是指脱氧核糖核酸(dna)、核糖核酸(rna)或其类似物。多核苷酸可以是单链的、双链的或含有单链和双链序列两者。多核苷酸分子可以来源于双链dna(dsdna)形式(例如,基因组dna、pcr和扩增产物等),或者可以来源于如dna(ssdna)或rna的单链形式并且其可以转化为dsdna形式,并且反之亦然。多核苷酸分子的准确序列可以是已知的或未知的。以下是多核苷酸的示例性实例:基因或基因片段(例如,探针、引物、est或sage标签)、基因组dna、基因组dna片段、外显子、内含子、信使rna(mrna)、转运rna、核糖体rna、核糖酶、cdna、重组多核苷酸、合成多核苷酸、分枝多核苷酸、质粒、载体、任何序列的分离的dna、任何序列的分离的rna、任何上述序列的核酸探针、引物或扩增拷贝。
144.多核苷酸可以包括核苷酸或核苷酸类似物。核苷酸通常含有糖、核碱基和至少一
个磷酸基。核苷酸可以是无碱基的(abasic)(即,缺少核碱基)。核苷酸包括脱氧核糖核苷酸、修饰的脱氧核糖核苷酸、核糖核苷酸、修饰的核糖核苷酸、肽核苷酸、修饰的肽核苷酸、修饰磷酸盐糖主链核苷酸及其混合物。核苷酸的实例包括(例如)腺苷一磷酸(amp)、腺苷二磷酸(adp)、腺苷三磷酸(atp)、胸苷一磷酸(tmp)、胸苷二磷酸(tdp)、胸苷三磷酸(ttp)、胞苷酸(cmp)、胞苷二磷酸(cdp)、胞苷三磷酸(ctp)、鸟苷一磷酸(gmp)、鸟苷二磷酸(gdp)、鸟苷三磷酸(gtp)、尿苷一磷酸(ump)、尿苷二磷酸(udp)、尿苷三磷酸(utp)、脱氧腺苷酸(damp)、脱氧腺苷二磷酸(dadp)、脱氧腺苷三磷酸(datp)、脱氧胸腺嘧啶核苷一磷酸(dtmp)、脱氧胸腺嘧啶核苷二磷酸(dtdp)、脱氧胸苷三磷酸(dttp)、去氧胞二磷(dcdp)、脱氧胞苷三磷酸(dctp)、脱氧鸟苷一磷酸(dgmp)、脱氧鸟苷二磷酸(dgdp)、脱氧鸟苷三磷酸(dgtp)、脱氧尿苷一磷酸(dump)、脱氧尿苷二磷酸(dudp)和脱氧尿苷三磷酸(dutp)。还可以在本文所述的方法中使用包含修饰的核碱基的核苷酸类似物。无论是具有天然主链还是类似结构,可以包含在多核苷酸中的示例性修饰的核碱基包括(例如)肌苷、黄嘌呤(xathanine)、次黄嘌呤(hypoxathanine)、异胞嘧啶、异鸟嘌呤、2
‑
氨基嘌呤、5
‑
甲基胞嘧啶、5
‑
羟甲基胞嘧啶、2
‑
氨基腺嘌呤、6
‑
甲基腺嘌呤、6
‑
甲基鸟嘌呤、2
‑
丙基鸟嘌呤、2
‑
丙基腺嘌呤、2
‑
硫脲嘧啶、2
‑
硫胸腺嘧啶、2
‑
硫胞嘧啶、15
‑
卤代脲嘧啶、15
‑
卤代胞嘧啶、5
‑
丙炔基尿嘧啶、5
‑
丙炔基胞嘧啶、6
‑
偶氮尿嘧啶、6
‑
偶氮胞嘧啶、6
‑
偶氮胸腺嘧啶、5
‑
尿嘧啶、4
‑
硫尿嘧啶、8
‑
卤代腺嘌呤或鸟嘌呤、8
‑
氨基腺嘌呤或鸟嘌呤、8
‑
硫腺嘌呤或鸟嘌呤、8
‑
硫烷基腺嘌呤或鸟嘌呤、8
‑
羟基腺嘌呤或鸟嘌呤、5
‑
卤素取代的尿嘧啶或胞嘧啶、7
‑
甲基鸟嘌呤、7
‑
甲基腺嘌呤、8
‑
氮杂鸟嘌呤、8
‑
氮杂腺嘌呤、7
‑
去氮鸟嘌呤、7
‑
去氮腺嘌呤、3
‑
去氮鸟嘌呤、3
‑
去氮腺嘌呤等。如本领域中已知的,某些核苷酸类似物不能引入多核苷酸,例如,核苷酸类似物,如腺苷5'
‑
磷酰硫酸。
145.如本文所使用的,术语“孔”旨在表示延伸穿过屏障(如膜)的结构,其允许离子和/或水溶性分子从屏障一侧穿过至屏障另一侧。孔可以(但不必)存在于膜中。例如,通常抑制离子或水溶性分子通过的屏障可以包括延伸穿过屏障以允许离子或水溶性分子从屏障一侧穿过至屏障另一侧的多孔结构。孔(例如,跨膜孔)包括(例如)生物孔、固态孔和生物和固态杂交孔。
146.如本文所使用的,术语“生物孔”旨在表示由生物来源材料制成的孔,其延伸穿过屏障(包括,例如,膜),其允许离子和/或水溶性分子从屏障一侧穿过至屏障另一侧。生物来源是指来源于或分离自生物环境(如生物或细胞)的材料,或生物学可用结构的合成产生形式。生物孔包括(例如)多肽孔和多核苷酸孔。
147.如本文所使用的,术语“多肽孔”旨在表示延伸穿过屏障(如,例如,膜)并且允许离子和/或水溶性分子从屏障一侧流至屏障另一侧的一个或多个多肽。多肽孔可以是单体、均聚物或杂聚物。多肽孔的结构包括(例如)α螺旋束孔和β
‑
折叠孔以及在本领域中熟知的所有其它结构。示例性多肽孔包括α
‑
溶血素、耻垢分枝杆菌(mycobacterium smegmatis)孔蛋白a、短杆菌肽a,麦芽糖孔蛋白、ompf、ompc、phoe、tsx、f
‑
pilus、sp1(wang等人,chem.commun.,49:1741
‑
1743,2013)和线粒体孔蛋白(vdac)xx、tom40(美国专利no.6,015,714和derrington等人,proc.natl.acad.sci.usa,107:16060(2010))。“耻垢分枝杆菌(mycobacterium smegmatis)孔蛋白a(mspa)”是分枝杆菌(mycobacteria)产生的膜孔蛋白,其允许亲水性分子进入细菌。mspa形成紧密互相连接的八聚物和类似于高脚杯的跨膜
β
‑
折叠,并且含有中性通道/孔。
148.如本文所使用的,术语“多核苷酸孔”旨在表示延伸穿过屏障(如,例如,膜)并且允许离子和/或水溶性分子从屏障一侧流至屏障另一侧的一个或多个多核苷酸。多核苷酸孔可以包括(例如)多核苷酸折纸。
149.如本文所使用的,术语“固态孔”旨在表示由非生物来源材料制成的孔,其延伸穿过屏障(如,例如,膜),并且允许离子和/或水溶性分子从屏障一侧穿过至屏障另一侧。固态旨在表示非生物来源的材料。固态孔可以是无机或有机材料。固态孔包括(例如)氮化硅孔、二氧化硅孔和石墨烯孔。
150.如本文所使用的,术语“生物和固态杂交孔”旨在表示由生物和非生物来源的材料两者制成的杂交孔,其延伸穿过屏障(如,例如,膜),并且允许水合离子和/或水溶性分子从屏障一侧穿过至屏障另一侧。生物来源的材料如上所定义并且包括(例如)多肽和多核苷酸。生物和固态杂交孔包括(例如)多肽
‑
固态杂交孔和多核苷酸
‑
固态孔。
151.如本文所使用的,术语“解旋酶”旨在表示具有使用来源于(例如)核苷三磷酸(ntp)水解的能量使双链多核苷酸打开的活性的多核苷酸结合蛋白。打开双链多核苷酸导致多核苷酸沿其活性位点易位。该术语旨在包含具有使单链多核苷酸以及部分双链多核苷酸易位或与之结合的活性的多肽。“hel308解旋酶”是atp依赖性dna解旋酶和超家族2解旋酶。来自果蝇(drosophila melanogaster)的组成成员mus308由融合至c末端dna聚合酶结构域的n末端sf2解旋酶结构域组成。智人中的hel308作为具有有限前行能力的3'至5'dna解旋酶sf2起作用。hel308解旋酶与ski2样解旋酶可互换使用。有用的同源物可以仅包含解旋酶结构域(即缺少聚合酶结构域)。仅有解旋酶的同源物存在于后生动物和古细菌中。后生动物的实例是人hel308和mus301。古细菌的实例是tga和mbu。
152.除非本文明确描述,否则如本文所使用的术语“hel308解旋酶底物”旨在表示核苷酸或核苷酸类似物,其能够被解旋酶水解并提供使双链或部分双链多核苷酸打开或使单链多核苷酸易位的能量。hel308解旋酶的常见底物包括atp。然而,该术语含义内的其它hel308解旋酶底物包括atp以外的核苷酸,如先前描述的那些,和能够被hel308解旋酶水解的核苷酸类似物。示例性类似物包括(例如)磷酸盐类似物,如γ巯基类似物、α巯基类似物等、atpγs、atpαs、amp、pnp、apcpp、appcp和appnhp。
153.如本文所使用的,术语“易位”旨在表示目标多核苷酸沿解旋酶和/或孔(或在它们内部)的运动。
154.如本文所使用的,当结合解旋酶使用时,术语“完整易位循环”旨在表示目标多核苷酸的一个或多个核苷酸单元沿解旋酶和/或孔运动的完整间隔。完全间隔可以在循环中的任一点开始,并且可以(例如)包括图3中所示的间隔,其包括atp结合以及所结合的atp水解的步骤。因此,如本文所使用的完整易位循环可以在核苷酸底物结合开始并在核苷酸底物水解结束。类似地,完整易位循环可以在核苷酸底物水解开始并在核苷酸结合结束。类似地,完整易位循环可以在以上举例说明的两个起始点之间的任一点开始,只要它在起始点前的步骤结束。
155.如本文所使用的,当结合解旋酶使用时,术语“分步易位步骤”旨在表示表征完整易位循环的一部分的可检测事件。例如,分步易位步骤可以是目标多核苷酸的一个或多个核苷酸单元沿解旋酶和/或孔的部分易位。在具体的实施方式中,当发生构象变化时,可以
symposium series 580,“carbohydrate modifications in antisense research”,y.s.sanghui和p.dan cook主编;mesmaeker等人,bioorganic&medicinal chem.lett.,4:395(1994);jeffs等人,j.biomolecular nmr,34:17(1994);tetrahedron lett.,37:743(1996))和非核糖主链的那些,所述非核糖主链包括美国专利no.5,235,033和5,034,506,以及第6和7章,asc symposium series 580,“carbohydrate modifications in antisense research”,y.s.sanghui主编p.dan cook中所述的那些。含有一个或多个碳环糖的多核苷酸分子也包含在多核苷酸的定义内(参见jenkins等人,chem.soc.rev.,(1995)第169
‑
176页)。在rawls,c&e news,jun.2,1997,第35页中描述了一些多核苷酸。
162.可以根据本发明所述的方法表征目标多核苷酸。示例性多核苷酸包括(例如)基因或基因片段(例如,探针、引物、est或sage标签)、基因组dna、基因组dna片段、外显子、内含子、信使rna(mrna)、转运rna、核糖体rna、核糖酶、cdna、重组多核苷酸、合成多核苷酸、分枝多核苷酸、质粒、载体、任何序列的分离的dna、任何序列的分离的rna、任何上述序列的核酸探针、引物或扩展拷贝。
163.在本文的具体实施方式中使用的目标多核苷酸可以具有任意多种长度,通常具有足以延伸通过孔并在孔的一侧被解旋酶结合的长度。一般说来,这种长度为至少约10个核苷酸长。然而,对于使用本发明所述的方法的表征来说,比该最小尺寸长的多个长度是适用的。有用的多核苷酸的示例性长度包括(例如)至少约10,20,30,40,50,60,70,80,90,100,150,200,300,400,500,1,000,5,000或10,000、100,000个核苷酸或更长。作为另外一种选择或者另外,所述长度可以不长于1,000,000、100,000、10,000、1,000、100个核苷酸或更少。因此,可以使用本发明所述的方法测序的多核苷酸可以(例如)在短多核苷酸、片段、cdna、基因和基因组片段的范围内。
164.在本发明所述的方法中使用的多核苷酸可以是单链的、双链的或含有单链和双链序列两者。多核苷酸分子可以来自于双链多核苷酸(例如,dsdna)并且可以转化为单链多核苷酸。多核苷酸分子还可以来自于单链多核苷酸(例如,ssdna、ssrna),并且所述ssdna可以转化为双链多核苷酸。在本发明公开的一些方面,双链或部分双链多核苷酸包括封闭性多核苷酸。这类多核苷酸可以包括结合本文中图13a
‑
13e、14a
‑
14d和15a
‑
15c举例说明的那些。在wo 2013/057495中描述了多核苷酸易位通过孔的示例性形式。
165.在一些方面,本发明公开提供了表征目标多核苷酸的方法。所述方法包括鉴别:(1)目标多核苷酸的序列;(2)目标多核苷酸的修饰;(3)目标多核苷酸的长度;(4)目标多核苷酸的身份;(5)目标多核苷酸的来源;或(6)目标多核苷酸的二级结构。
166.多核苷酸序列是指多核苷酸的一级结构或者多核苷酸分子中核苷酸的相继次序。通过使用目标多核苷酸通过孔的分步易位步骤所产生的信号来表征目标多核苷酸中的核苷酸,可以确定多核苷酸的序列。
167.多核苷酸的修饰是指多核苷酸中核苷酸的任何共价或非共价修饰,包括(例如)核苷酸甲基化或羟甲基化。的确,修饰可以包括可以引入多核苷酸单链中的任意数目的核苷酸类似物,包括,例如,8
‑
氧代鸟苷、5
‑
甲酰胞嘧啶和5
‑
羧基胞嘧啶以及在本文其它处所描述的其它类似物。核苷酸的修饰提供了信号中相应的变化。因此,通过使用目标多核苷酸通过孔的分步易位步骤所产生的信号来表征目标多核苷酸中修饰的核苷酸,可以确定多核苷酸的一个或多个修饰。
168.多核苷酸的长度是指多核苷酸中核苷酸的数目。多核苷酸的长度可以通过(例如)确定多核苷酸的一级序列或通过测量其在孔中的停留时间或者通过对穿过孔的核苷酸数目计数来确定。在一些实施方式中,停留时间对应于电流瞬时变化的持续时间。瞬时变化可以认为是由于多核苷酸的存在所造成的孔电流的任何偏离。在一些实施方式中,所述偏离导致电流幅度减小。这种减小通常可以是原始未封闭的孔电流的至多95%,90%,80%,60%,50%,40%,30%,20%或10%或更小。作为另外一种选择或者另外,所述减小可以是至少10%,20%,30%,40%,50%,60%,70%,80%或90%或以上。在一些情况下,多核苷酸可以导致电流幅度相对于未封闭孔提高。可以通过取决于所使用的实验条件的可重复数学函数来描述持续时间和多核苷酸长度之间的关系。对于给定类型的多核苷酸(例如,dna或rna),所述函数可以是线性或非线性的(例如,s形或指数)函数。
169.多核苷酸的身份是指多核苷酸的类型。身份还可以是指如本领域中已知的多核苷酸的名称。例如,多核苷酸的身份可以是(例如)dna、rna、双链多核苷酸、单链多核苷酸和/或部分双链多核苷酸。多核苷酸的身份还可以包括确定多核苷酸的基因产物或结构功能。例如,多核苷酸可以编码多肽或者它可以是结构多核苷酸,如核糖体rna。可以根据所有或部分多核苷酸的核苷酸序列、与所有或部分多核苷酸互补的第二多核苷酸的序列、所有或部分多核苷酸所编码的rna序列或者所有或部分多核苷酸所编码的蛋白质序列来确定多核苷酸的身份。在具体的实例中,可以通过构成多核苷酸一部分的“标签”或“条形码”序列鉴别多核苷酸。在这些实例中,可以通过对标签或条型码所预期的信号类型来分配多核苷酸的身份。多核苷酸的来源可以是指多核苷酸的物种来源或是指合成来源。可以使用本领域中熟知的程序,例如,blastn,通过在多核苷酸序列数据库中对多核苷酸序列进行比对来确定多核苷酸的身份和来源。
170.多核苷酸的二级结构是指多核苷酸分子中自互补性区域的分子内碱基配对。示例性二级结构包括(例如)双螺旋、发夹、环、凸起(bulges)、双螺旋、接合(junction)、茎、假结(pseudoknot)、三螺旋、h
‑
dna、锤头型(hammerhead)和自拼接核糖酶。可以(例如)通过测量其在孔中停留时间的相应变化或测量通过分步易位步骤产生的信号的相应变化来确定多核苷酸的二级结构。
171.孔是延伸穿过屏障(包括,例如,膜)的结构,其允许离子和/或水溶性分子从屏障一侧穿过至屏障另一侧。孔可以(但不必)存在于膜中。例如,通常抑制离子或水溶性分子通过的屏障可以包括延伸穿过屏障以允许离子或水溶性分子从屏障一侧穿过至屏障另一侧的多孔结构。本发明公开的膜可以是(例如)不透性或半透性屏障,其分成可以具有相同或不同的组成的两个液体室。可以根据本发明公开使用任何膜,只要所述膜可以配置以包含跨膜的孔并维持膜两侧的势差。适合的势差如下所述。
172.可以在本发明公开的组合物和方法中使用本领域中熟知的多种膜。在本领域中熟知的这些膜包括多种不同的结构和组成。例如,膜可以是单层或多层结构,只要可以引入用于多核苷酸表征的孔。膜中的层是指形成屏障的不透性或半透性材料。以下还描述了单层和多层膜的实例。
173.形成膜的材料可以是生物或非生物来源的。生物来源的材料是指来源于或分离自生物环境(如生物或细胞)的材料,或生物学可用结构的合成产生形式。由生物来源材料制成的示例性膜包括脂质双分子层。非生物来源的材料也称为固态材料并且可以形成固态
膜。
174.适合的脂质双分子层和制备或获得脂质双分子层的方法在本领域中是熟知的并且在(例如)美国专利公开us 2010/0196203和pct专利公开wo2006/100484中公开。适合的脂质双分子层包括(例如)细胞膜、细胞器膜、脂质体、平面脂质双分子层和支撑的脂质双分子层。可以(例如)从两个相对的磷脂层形成脂质双分子层,所述磷脂的布置使得它们疏水性尾部基团彼此面对以形成疏水性内部,而脂肪的亲水性头部基团朝外面对双分子层每侧的水相环境。还可以(例如)通过montal and mueller的方法(proc.natl.acad.sci.usa.,1972;69:3561
‑
3566)形成脂质双分子层,其中脂质单分子层从水溶液/空气界面上通过垂直于界面的孔的任一侧。通常通过首先将其溶于有机溶剂,然后使一滴溶剂在孔任一侧上的水溶液表面上蒸发,将脂质加入到电解质水溶液的表面。一旦有机溶剂蒸发,孔任一侧上的溶液/空气界面物理地上下移动通过孔直至形成双层。双分子层形成的其它常规方法包括尖端浸渍(tip
‑
dipping)、涂覆双分子层(painting bilayers)和脂质体双分子层膜片钳(patch
‑
clamping)。获得或产生脂质双分子层的多种其它方法在本领域中是熟知的并且等同地适用于本发明公开的组合物和方法中的用途。
175.固态膜在本领域中是熟知的并且在(例如)pct专利公开wo 2000/079257中公开。如上所述,固态膜由非生物来源的材料的一个或多个层制成。固态膜可以是单层,如支持基底上的涂层或膜,或者是自立式成分(free
‑
standing element)。固态膜还可以是夹心构造的多层复合材料。对根据本发明公开可以使用的材料没有具体限制,只要可以配制所得固态膜以包含跨膜孔并且在膜两侧建立势差。可以从有机和无机材料制备固态膜,包括(例如)微电子材料、绝缘材料如si3n4、al2o3和sio、有机和无机聚合物如聚酰胺、三嵌段共聚物(例如,两亲pmoxa
‑
pdms
‑
pmoxa aba三嵌段共聚物)、塑料材料如或弹性体如二组分加成
‑
固化硅橡胶和玻璃。另外,可以从石墨烯单层(石墨烯是密集挤压成二维蜂房晶格的原子薄层)、石墨烯多层或与一个或多个其它固态材料层混合的一个或多个石墨烯层制备固态膜(pct专利公开wo 2013/016486)。含有石墨烯的固态膜可以包括石墨烯纳米带或石墨烯纳米间隙中的至少一种石墨烯层,其可以用作电学传感器来表征目标多核苷酸(参见pct专利公开wo 2013/016486)。可以通过本领域中熟知的方法制备固态膜。例如,可以通过化学气相淀积(cvd)或从石墨剥落(pct专利公开wo 2013/016486)制备石墨烯膜。
176.本发明公开的组合物和方法可以使用位于屏障中的孔以用于目标多核苷酸的表征。可以从生物或非生物来源的材料制备孔。因此,孔包括(例如)生物孔、固态孔和生物和固态杂交孔。
177.孔可以具有有利于多核苷酸中核苷酸序列检测的与其相关的功能性。例如,孔可以包含酶(如解旋酶)或者其它功能性,所述酶或其它功能性连接至孔、与孔结合或位于孔附近以控制多核苷酸通过孔的速率。孔可以具有与之结合的检测电路或传感器,包括(例如)膜片钳电路、隧道电极电路或横向电导测定电路(如石墨烯纳米带或石墨烯纳米间隙)。孔还可以包括检测多核苷酸上标记物(包括(例如)荧光部分或拉曼信号产生部分)的光学传感器,其基于片段与孔(例如,将片段通过孔)的相互作用确定核苷酸序列。
178.在具体的实施方式中,可以在本发明公开的组合物和方法中使用生物孔(包括多肽孔和多核苷酸孔),只要所述孔具有允许多核苷酸通过屏障(例如,膜)的缢缩区。缢缩区是孔腔中的位置,在此分析物(例如,多核苷酸或核苷酸)的阻塞影响了孔所产生的可检测
信号。可以在本发明公开的组合物和方法中使用具有多个缢缩区长度的孔,其包括(例如)至少1、2、3、4、5、6、7、8、9或10个核苷酸的长度。作为另外一种选择或者另外,可以使用至多约10、9、8、7、6、5、4、3、2或1个核苷酸长度。然而,缢缩区长度可以影响信号质量。例如,较短的缢缩区可以导致更好的核苷酸易位分辨率或重构准确度。在一个实施方式中,生物孔具有约5个核苷酸或以下的缢缩区,位于缢缩区的5个或小于5个核苷酸调节电信号,其具有比得自大于5个核苷酸的电信号更好的核苷酸易位分辨率。在一些情况下,对于小于2nt的缢缩,信噪比提高不会导致测序准确度改善。当由于酶的随机运动而跳过核苷酸时,如果大于较小缢缩的均聚物不再能被检测并且再读取的缺少降低准确度时,这可以产生。因此,根据本发明公开,可以使用适合的多肽孔和具有5个核苷酸或以下的缢缩区的多核苷酸孔。考虑到本文所提供的教导和指导,本领域技术人员将理解对于具体需要,什么长度的缢缩区是适用的。例如,在需要高质量结果的应用中,本领域技术人员可以使用具有较短缢缩区的孔。
179.生物孔是由生物来源材料制成,延伸穿过屏障(例如,膜)的孔,其允许离子和/或水溶性分子从屏障一侧穿过至屏障另一侧。如本文所述使用的膜,当提及孔时,生物来源是指来源于或分离自生物环境(如生物或细胞)的结构,或者生物学可用结构的合成产生形式。生物来源材料包括(例如)多肽和多核苷酸。因此,生物孔包括(例如)多肽孔和多核苷酸孔。
180.重构成屏障(例如,膜),如脂质双分子层的多肽孔可以用于纳米孔测序。存在可以根据本发明公开使用的多种多肽孔,只要多肽可以形成允许目标多核苷酸穿过屏障(例如,膜)的缢缩区。根据所涉及的多肽,多肽孔可以是单体、均聚物或杂聚物。多肽孔可以包括一些重复亚基,如7或8个亚基。因此,所述多肽孔可以是(例如)六聚、七聚或八聚孔。
181.多肽孔包括(例如)α
‑
螺旋束孔和β
‑
折叠孔以及在本领域中熟知的所有其它孔。α
‑
螺旋束孔包括由α
‑
螺旋形成的孔。适合的α
‑
螺旋束孔包括(例如)内膜蛋白和α外膜蛋白,如wza和clya毒素。β
‑
折叠孔包括通过β
‑
链形成的孔。适合的β
‑
折叠孔包括(例如)β
‑
毒素,如α
‑
溶血素、炭疽毒素和杀白细胞素,和细菌的外膜蛋白/孔蛋白,如耻垢分枝杆菌(mycobacterium smegmatis)孔蛋白(msp),包括mspa、外膜孔蛋白f(ompf)、外膜孔蛋白g(ompg)、外膜磷脂酶a和奈氏球菌(neisseria)自转运蛋白脂蛋白(nalp)。其它孔包括(例如)胞溶素(参见,例如,wo 2013 153359)或来自皮疽诺卡氏菌(norcadia farcinica)的mspa同源物。
182.α
‑
溶血素多肽是可以在本发明公开的方法和组合物中使用的七聚多肽孔。它由连接到5nm长的β
‑
折叠的3.6nm孔腔(vestibule)组成,其含有允许单链多核苷酸通过但不允许双链多核苷酸通过的1.4nm缢缩。α
‑
溶血素的约5nm长的圆柱形β
‑
折叠孔每次可以容纳多至约10个核苷酸。位于该β
‑
折叠中的核苷酸显著调节孔电流并随后削弱对最窄1.4nm的孔缢缩中的单个核苷酸特异的离子特征,从而降低测序应用中核苷酸易位的整体分辨率。
183.mspa是可以在本发明公开的组合物和方法中使用的八聚多肽孔。它含有直径~1.2nm,缢缩长度~0.5nm的单个缢缩;与α
‑
溶血素的圆柱形结构相反,内孔形成锥形漏斗形状。derrington等人表明基因工程mspa区分三核苷酸组(aaa、ggg、ttt、ccc)的能力,其核苷酸分离效率与天然α
‑
溶血素相比具有令人深刻印象的3.5倍提高(derrington等人,proc.natl.acad.sci.usa,107:16060(2010))。据报导在涉及固定化单链多核苷酸的实验
中,观察到mspa缢缩内或附近少至3个核苷酸有助于孔电流,比已知调节天然α
‑
溶血素中离子电流的~10个核苷酸具有显著改善。作者假设这可以通过定点突变进一步改善至或许单个核苷酸,这是未来mspa突变的目标。
184.在一些方面,多肽孔为耻垢分枝杆菌(mycobacterium smegmatis)孔蛋白a(mspa)。在一些方面,所述mspa具有seq id no:1所示或与seq id no:1具有至少15%、至少20%、至少25%、至少30%、至少35%、至少40%、至少45%、至少50%、至少55%、至少60%、至少65%、至少70%、至少75%、至少80%、至少85%、至少90%、至少95%或至少99%同源性的氨基酸序列。
185.mspa是适合的多肽孔。另外,mspa突变体可以在本发明公开的组合物和方法中使用以调节多核苷酸通过孔的易位。在本文的实施方式中使用的mspa孔可以具有seq id no:1所示的氨基酸序列,其对应于gldnelslvdgqdrtltvqqwdtflngvfpldrnrltrewfhsgrakyivagpgadefegtlelgyqigfpwslgvginfsyttpnilinngnitappfglnsvitpnlfpgvsisarlgngpgiqevatfsvrvsgakggvavsnahgtvtgaaggvllrpfarliastgdsvttygepwnmn,它是具有以下突变的mspa序列:d90n、d91n、d93n、d118r、d134r&e139k。seq id no:1所示的mspa孔突变体被称为“m2 nnn”。可以在本发明公开的组合物和方法中使用其它mspa突变体,其与seq id no:1具有至少15%、至少20%、至少25%、至少30%、至少35%、至少40%、至少45%、至少50%、至少55%、至少60%、至少65%或至少70%、至少75%、至少80%、至少85%、至少90%、至少95%或至少99%同源性。对另一序列具有特定百分比(例如,50%)的同源性的多肽或多肽区(或者多核苷酸或多核苷酸区)是指当比对时,相比较的两个序列中氨基酸(或核苷酸碱基)的百分比是相同的。如本文所述,可以使用本领域中已知的软件程序来进行确定它们序列同一性百分比的两个序列的比对。可以根据本领域熟知的方法对天然mspa多肽进行突变,包括特定区域或特定氨基酸残基的插入、缺失、取代或其它所选修饰,所述突变方法包括编码mspa多肽的核酸的定点突变(zoller,m.j.,curr.opin.biotechnol.,3:348
‑
354,(1992))。在us2012/0055792a1中还描述了有用的mspa突变体。
186.可以通过多种本领域熟知的方法分离在本发明公开的组合物和方法中使用的天然或突变mspa多肽,例如,重组表达系统、沉淀、凝胶过滤、离子交换、反相和亲和色谱法等。其它熟知的方法描述于deutscher等人,guide to protein purification:methods in enzymology,vol.182,(academic press,(1990))。作为另外一种选择,可以使用熟知的重组方法获得本发明公开的分离的天然或突变mspa多肽。本领域技术人员可以选择本发明公开的天然或突变mspa多肽的生物纯化的方法和条件,并且可以(例如)通过功能测定来监控纯化。
187.制备天然或突变mspa多肽的一个示例性方法是使用本领域中熟知的方法在适合的宿主细胞,如细菌细胞、酵母细胞或其它适合的细胞中表达编码所述多肽的多核苷酸,并回收所表达的天然或突变体mspa多肽,再使用熟知的纯化方法,如本文所述的那些。可以直接从已用如本文所述的表达载体转化的细胞中分离天然或突变mspa多肽。还可以将重组表达的天然或突变mspa多肽表达为具有适当亲和标签的融合多肽,所述标签如谷胱甘肽s转移酶(gst)或多组氨酸,并亲和纯化。还可以使用本领域技术人员熟知的多肽合成方法,通过化学合成产生天然或突变mspa多肽。
188.重构成屏障(例如,膜),如脂质双分子层的多核苷酸孔还可以用于纳米孔测序。多
核苷酸孔是延伸穿过屏障(例如,膜)并允许离子和/或水溶性分子从屏障一侧穿过至屏障另一侧的一个或多个多核苷酸。可以根据本发明公开的具体实施方式使用任何多核苷酸孔,只要多核苷酸可以形成允许目标多肽穿过屏障(例如,膜)的缢缩区。示例性多核苷酸孔包括(例如)多核苷酸折纸孔(polynucleotide origami pore)。可以使用“折纸法”制备图形在二维或三维空间中延伸的多核苷酸折纸孔,如rothemund,nature,440:297
‑
302(2006)中所述。折纸是利用基因组多核苷酸的长链和多个较短的合成“订书针”多核苷酸链产生延伸结构的通用技术。最初的折纸结构基本上是二维结构。此后,折纸技术扩展至三维结构(douglas等人,nature 459:414
‑
418(2009);ke等人,nano letters,6:2445
‑
2447(2009);andersen等人,nature 459:73
‑
76(2009))。
189.在本发明公开的组合物和方法中还可以使用固态孔。固态孔是由非生物来源的材料制成的孔,其延伸穿过屏障(例如,膜),允许离子和/或水溶性分子从屏障一侧穿过至屏障另一侧。
190.可以通过在固态屏障(例如,膜)中产生孔来形成固态孔。因此,与固态膜类似并且如本文所述,可以通过多种材料形成固态孔,包括无机和有机材料两者。
191.适合的固态孔包括(例如)氧化铝、氧化钽、氧化钛、二氧化硅、二氧化铪、氧化锆、氮化硼、氮化硅、石墨烯或其纳米层合物(例如,石墨烯
‑
al2o3)或它们的任意组合(pct专利公开wo 2013016486a1)。可以通过使用定制的反馈控制离子束雕刻工具或使用来自场发射枪(feg)tem的聚焦汇聚电子束在膜中分解溅射(decompositionally sputter)纳米孔或本领域中熟知的任何其它方法来制备固态孔(pct专利公开wo 2013016486a1)。例如,可以通过使用来自feg tem的聚焦汇聚电子束钻穿石墨烯
‑
al2o3膜制备石墨烯纳米层合物孔,如石墨烯
‑
al2o3孔(venkatesan等人,acs nano.,6:441
–
450(2012))。
192.可以在本发明公开的组合物和方法中使用生物和固态杂交孔。生物和固态杂交孔是由生物来源和非生物来源的材料制成的杂交孔,其延伸穿过屏障(例如,膜),允许离子和/或水溶性分子从屏障一侧穿过至屏障另一侧。生物来源的材料如上所定义并且包括(例如)多肽和多核苷酸。非生物来源的材料被称为固态材料,如本文所述。
193.因此,生物和固态杂交孔包括(例如)多肽
‑
固态杂交孔和多核苷酸
‑
固态杂交孔。多肽
‑
固态杂交孔包括一种或多种多肽和固态材料。多核苷酸
‑
固态杂交孔包括一种或多种多核苷酸和固态材料。通过将多肽或多核苷酸孔与固态孔工程设计来制备生物和固态杂交孔(参见pct专利公开wo 2013/016486)。以上描述了适合的多肽孔、多核苷酸孔和固态孔的实例。
194.纳米孔测序装置可以具有单孔或多孔。多孔可以作为纳米孔阵列使用以表征不止一个具有相同或不同组成的目标核苷酸。本文所使用的多孔的示例性数目包括(例如)至少1、4、16、64、256、512、1028、4096、16384、32768、100000、100万、1000万个孔或以上。在优选的实施方式中,多孔的数目将大于4096。纳米孔阵列在本领域中是已知的并且在(例如)pct专利公开wo 2013/016486中公开。例如,可以使用电子束平印术和反应性离子蚀刻步骤在sin/al2o3膜中制造~15nm直径的固态孔的高密度阵列,从而有利于多核苷酸分子的高通量分析。
195.本发明所述的方法可以使用跨越屏障(例如,膜)的势差。所述势差可以是势差、化学势差或电化学势差。可以通过向至少一个液池注入或施用电流的电压电源将势差加载到
屏障(例如,膜)两侧。可以通过双池的离子组成的差异,将化学势加载到屏障两侧。可以通过双池离子组成的差异并结合电势来建立电化学势差。不同的离子组成可以是(例如)每个池中不同的离子或者每个池中相同离子的不同浓度。
196.将电势应用于孔两侧以迫使多核苷酸易位通过孔在本领域中是熟知的并且可以根据本发明公开使用(deamer等人,trends biotechnol.,18:147
‑
151(2000);deamer等人,ace chem res.,35:817
‑
825(2002);和li等人,nat mater.,2(9):611
‑
615(2003))。可以通过应用于孔两侧的电压实施本发明所述的方法。电压范围可以选自40mv至1v以上。通常,本发明所述的方法将在100至200mv的范围内进行。在具体的实例中,所述方法在140mv或180mv进行。在马达运动期间,电压不需要是稳定的。通常应用电压极性,从而通过电泳驱使带负电荷的多核苷酸进入孔中。在一些情况下,可以降低电压或反转极性以有利于马达的适当功能。
197.在一些情况下,可以使用压力差的应用以迫使多核苷酸易位通过孔。可以在本文举例说明的方法中电势或其它势差的位置使用压力差。
198.本发明所述的方法生产了对应于一个或多个核苷酸通过孔的易位的一个或多个信号。因此,例如,当目标多核苷酸通过孔时,由于缢缩的碱基依赖性阻塞,穿过屏障的电流改变。可以使用如本文所述的或本领域中已知的任何多种方法测量来自电流变化的信号。如上所述,每个信号对孔中的核苷酸物质是唯一的,从而所得信号可以用于确定多核苷酸的特征。例如,可以确定产生特征信号的一种或多种核苷酸物质的身份。在本发明所述的方法中有用的信号包括(例如)电信号和光信号,以下将进一步描述。在一些方面,电信号可以是电流、电压、隧穿、电阻、电压、电导率的测量;或者横向电测量(pct专利公开wo 2013/016486)。在一些方面,电信号是通过孔的电流。
199.在本文所述方法中检测的电信号可以是流动通过孔的电流,它是电荷流(deamer等人,trends biotechnol.,18:147
‑
151(2000);deamer等人,ace chem res.,35:817
‑
825(2002);和li等人,nat mater.,2(9):611
‑
615(2003))。如本文所述,可以使用与孔偶联的检测电路测量电信号,例如,膜片钳电路或隧道电极电路。可以检测的电压、隧穿、电阻和电导率信号的实例和用于它们检测的装置在本领域中是已知的,如(例如)wanunu,phys life rev.,9(2):125
‑
58(2012);和venkatesan等人,nat nanotechnol.,6(10):615
‑
24(2011)中所述。
200.在本发明所述的方法中有用的光信号包括(例如)荧光信号和拉曼信号。可以通过将目标核苷酸与光信号产生标记物(例如,荧光部分或拉曼信号产生部分)偶联来产生光信号。例如,在dela torre等人,nanotechnology,23(38):385308(2012)中,使用全内反射荧光(tirf)显微术的光学方案来照射宽的tio2
‑
涂覆膜区域。在soni等人,rev sci instrum.,81(1):014301(2010)中,使用方法将两个单分子测量方式结合,即全内反射显微术和使用纳米孔的生物分子电学检测。
201.如本文所述,可以将孔与传感电路偶联,包括(例如)膜片钳电路、隧道电极电路或横向电导测定电路(如石墨烯纳米带或石墨烯纳米间隙)以记录本发明实施方式中的电信号。另外,还可以将孔与检测多核苷酸上的标记物,例如,荧光部分或拉曼信号产生部分的光学传感器偶联。
202.纳米孔测序方法可以使用减缓目标多核苷酸通过孔的易位的机制。例如,可以连
接或掺入多核苷酸结合蛋白,如解旋酶、易位酶或聚合酶以调节易位速率。所述连接可以是(例如)瞬时的或持久的,并且可以通过目标多核苷酸(由于将其从孔中拉出)或者通过在本领域中熟知的多种多肽、化学接头或捕获部分介导。示例性技术描述于manrao等人,nat biotechnol.,30(4):349
–
353(2012)和cherf等人,nat biotechnol.,30(4):344
–
348(2102)。在具体的实施方式中,解旋酶或其它分子马达可以用于减缓或停止目标多核苷酸通过孔的易位。例如,当使用水解核苷酸以影响易位的马达时,可以从马达漏去核苷酸和/或可以使马达经受抑制剂(例如,不可水解的核苷酸类似物),从而目标多核苷酸仍与马达结合并且不明显易位通过孔。在一些实施方式中,随后可以通过将核苷酸递送至马达和/或除去抑制剂来允许易位发生。本发明所述的方法可以包括将孔与目标多核苷酸和hel308解旋酶接触的步骤以控制多核苷酸通过孔的易位速率。如以下进一步描述的,hel308解旋酶可以鉴别为atp
‑
依赖性dna解旋酶和超家族2解旋酶。考虑到本文所提供的教导和指导,对于根据本发明的实施方式的用途,本领域技术人员可以适合地选择或改变任何hel308解旋酶。以下进一步描述了适合的hel308解旋酶。
203.在本文所述方法的一些方面,目标多核苷酸的易位处于通过孔的电流方向的相反方向。在其它方面,目标多核苷酸的易位处于通过孔的电流方向的相同方向。
204.因此,可以在至少两个模式中进行本发明所述的方法,其中目标多核苷酸的易位与(例如)通过孔的电流或其它势的方向相反或相同。该结果可以通过将本发明公开的hel308解旋酶与目标多核苷酸的5'或3'端结合来实现。当提及双链多核苷酸时,5'或3'取向是指双链多核苷酸内的单链。因此,hel308解旋酶可以将多核苷酸从孔中拉出或送入孔中,即与电压梯度对多核苷酸产生的作用力的方向相反(参见图13a
‑
13e和14a
‑
14d),或者当多核苷酸在与电压梯度或其它势所产生的作用力相同的方向移动时,使用解旋酶调节易位速度(参见图15a
‑
15c)。
205.图13a
‑
13e举例说明了根据一些实施方式,针对势(如电压梯度)所产生的作用力,hel308解旋酶控制的多核苷酸易位,例如,基于具有hel308解旋酶3'突出结合位点和胆固醇双分子层锚点的三元多核苷酸复合物。实心圆环(
●
)表示5'磷酸盐。实心菱形()表示3'胆固醇。具有凹口的实心半透明圆表示hel308解旋酶。虚线表示任意长度。大的灰色箭头表示多核苷酸进入或移出孔的多核苷酸移动的方向(通过或抵抗外加场)。大的黑色箭头表示解旋酶沿多核苷酸的易位方向,其为3'至5’。孔(漏斗状圆锥形物体)位于膜(双水平线)中。
206.图13a
‑
13e示出了具有hel308解旋酶3'突出结合位点和胆固醇双分子层锚点的三元多核苷酸复合物用于多核苷酸序列的使用。胆固醇
‑
标记的多核苷酸“i”是任选的并且用于杂交至目标多核苷酸“ii”,目标多核苷酸“ii”将易位通过孔并且有利于整个复合物向脂质双分子层的招募(图13a)。例如,通过电压梯度将5'磷酸盐拉动穿过孔,从而导致目标多核苷酸“ii”的5'端首先进入孔,并导致胆固醇
‑
标记的多核苷酸剥落(图13b)。随着含有磷酸盐的多核苷酸被拉动通过孔至相反一侧,第二杂交多核苷酸“iii”剥落,这是因为孔过窄从而不允许双链多核苷酸易位(13c)。多核苷酸iii的一个目的是产生hel308解旋酶可以优先结合的hel308解旋酶结合位点,它通常是约8个核苷酸的3'单链多核苷酸突出。此外,通过迫使hel308解旋酶分子在易位多核苷酸的3'端结合,将易位通过孔的多核苷酸的长度最大化。复合物的多核苷酸“iii”可以是任何长度,包括任意长度,并且3'端不需要临近多核苷酸“i”的5'端。一旦到达孔口,hel308解旋酶通过其3'至5'易位酶活性,将多核苷酸逆电
压梯度反向拉入顺室(cis chamber)(图13d和13e)。
207.图14a
‑
14d还示出了根据一些实施方式,针对势(如电压梯度)所产生的作用力,hel308解旋酶控制的多核苷酸易位,例如,基于具有hel308解旋酶3'突出结合位点和胆固醇双分子层锚点的三元多核苷酸复合物。实心圆环(
●
)表示5'磷酸盐。实心菱形()表示3'胆固醇。具有凹口的实心半透明圆表示hel308解旋酶。虚线表示任意长度。大的灰色箭头表示多核苷酸进入或移出孔的多核苷酸移动的方向(通过或抵抗外加场)。大的黑色箭头表示解旋酶沿多核苷酸的易位方向,其为3'至5’。孔(漏斗状圆锥形物体)位于膜(双水平线)中。然而,该方案举例说明了使用单个杂交多核苷酸“i”在目标多核苷酸“ii”上产生用于hel308解旋酶结合的3'突出,并且还含有任选的胆固醇部分。有可能hel308解旋酶可以结合多核苷酸“ii”的单链区上的任何位置。示出了多个hel308解旋酶分子,并通过“e1”、“e2”和“e3”表示。到达孔口的解旋酶首先将引起返回同侧(cis side)的控制易位过程。如果它脱离,则将发生不受控制的易位直至下一个结合的hel308解旋酶分子到达孔口并开始控制的易位。
208.图15a
‑
15c举例说明了当多核苷酸在与势(如电压梯度)所产生的作用力相同的方向移动时,使用hel308解旋酶调节多核苷酸易位速度。具有凹口的实心半透明圆表示hel308解旋酶。虚线表示任意长度。大的灰色箭头表示通过外加场多核苷酸移动进入孔的方向。大的黑色箭头表示解旋酶沿多核苷酸的易位方向,其为3'至5’。孔(漏斗状圆锥形物体)位于膜(双水平线)中。在该示例性方案中,目标多核苷酸的3'端首先进入孔。当沿易位多核苷酸的3'至5'端易位时,hel308解旋酶控制多核苷酸的易位速度。
209.如上所述,在hel308解旋酶的背景中,分步易位步骤可以表示目标多核苷酸的一个或多个核苷酸沿解旋酶和/或孔的部分易位。因此,分步易位步骤是指小于完整易位循环的核苷酸步骤的一部分。当发生构象变化时,在atp结合和水解之间发生分步易位步骤。完整核苷酸步骤可能需要一个或多个分步易位步骤。构象变化有效地将完整易位循环划分为至少两个部分或分步易位步骤。
210.可以以相同方式使用部分或分步易位步骤以产生表征通过孔的一个或多个核苷酸的唯一信号。因此,由于对于易位通过孔的一个或多个核苷酸的每一个,电流变化对应于每个分步易位步骤,因此本发明所述的方法可以产生至少两个电信号。因此,在一些方面,分步易位步骤包括hel308解旋酶完整易位循环的第一分步易位步骤。在其它方面,分步易位步骤包括hel308解旋酶完整易位循环的第二分步易位步骤。可以单独或与其伴侣(例如,分别为第二或第一分步易位步骤)一起使用第一或第二分步易位步骤中的每一个以表征通过孔缢缩区的一个或多个核苷酸。
211.例如,如实施例i中进一步描述的,hel308解旋酶可以结合至atp并经历构象变化,从而提供第一分步易位步骤,并且hel308解旋酶可以通过atp水解将目标多核苷酸的一个或多个核苷酸沿解旋酶和/或孔易位,从而提供第二分步易位步骤。可以使用第一和第二分步易位步骤中的任一个或两者来确定(例如)产生信号的核苷酸或一个或多个核苷酸的核苷酸序列。当通过不止一个核苷酸产生信号时,则产生信号的多核苷酸部分被称为单词。因此,该类核苷酸单词可以是至少4、5、6、7、8、9、10个或更多个核苷酸长度并且对应于孔缢缩区的长度。作为另外一种选择或另外,核苷酸单词可以是至多10、9、8、7、6、5或4个或更少的核苷酸长度。
212.如上所述和如以下实施例iii中进一步举例说明的,可以使用得自完整易位循环的两个部分步骤的电信号鉴别多核苷酸中一个或多个核苷酸残基。使用来自两个分步易位步骤的信号对相同的一个或多个核苷酸提供了重复信号并使得能够在单个确定内获得更大的准确度。因此,使用来自两个部分易位步骤的信号可以导致表征准确度提高,与使用得自完整易位循环的单个电信号或其它信号的一个或多个核苷酸的表征相比,其差错率降低了25至50%。类似地,使用来自部分易位步骤的信号可以导致差错率降低至少5%、10%、20%、30%、40%、45%、55%、60%、65%、70%、75%、80%、85%、90%或更高。考虑到本文所提供的教导和指导,本领域技术人员将知道出于给定目的如何调节准确度,如通过(例如)降低如上所述的缢缩区的尺寸来提高核苷酸易位的分辨率。
213.在其它实施方式中,得自分步易位步骤的其它信息可以用于在许多方面提高纳米孔测序。例如,对于相同核苷酸单词,可以在算法中使用得自分步易位步骤的测量以改善纳米孔碱基调用准确度。对于相同的核苷酸单词,得自分步易位步骤的测量可以用于降低均聚物读取差错率,这是因为在单次确定中对相同核苷酸单词读取了两次。因此,对于相同核苷酸单词,得自分步易位步骤的测量使天然多核苷酸易位反应的可分辨的分辨率加倍,从而导致序列特异性图形的分辨率提高。后者的一个使用是用于检测序列重复或单核苷酸多态性(snp)的序列特异性图形识别算法。
214.如上所述,方法可以包括(a)跨越与hel308解旋酶和目标多核苷酸接触的孔施加势差;(b)测量目标多核苷酸通过孔的一个或多个分步易位步骤产生的一个或多个信号;和(c)根据分步易位步骤的电信号,表征目标多核苷酸。在一些方面,所述方法还包括重复步骤(a)
‑
(c)一次。通过重复步骤(a)
‑
(c),可以表征相邻核苷酸或相邻核苷酸单词。可以根据需要重复步骤(a)
‑
(c)直至表征了一些或所有目标多核苷酸。例如,可以通过步骤(a)
‑
(c)的任何所需迭代次数确定目标多核苷酸的部分或全部的序列。因此,可以确定目标多核苷酸的整体或部分的一个或多个特征。
215.如本文所述,根据本发明的实施方式,可以使用任何hel308解旋酶或其变体。下表1和2中提供了示例性hel308解旋酶。
216.表1.示例性hel308解旋酶
217.218.219.220.221.222.223.224.225.226.227.228.229.230.231.232.233.[0234][0235]
下表2示出了hel308解旋酶以及hel308基序和扩展的hel308基序的更多实施方式。
[0236]
表2.示例性hel308解旋酶、hel308基序和扩展的hel308基序。
[0237]
[0238]
[0239][0240]
**:该hel308解旋酶序列的相关详细信息,参见国际专利公开no.wo2013/057495。
[0241]
在本发明实施方式中,还可以使用保留了多核苷酸结合和解旋酶酶活性的hel308解旋酶的变体或突变体。可以根据本领域熟知的方法获得这种变体或突变体,包括编码天然hel308解旋酶的核酸的定点突变(zoller,m.j.,curr.opin.biotechnol.,3:348
‑
354,(1992))。
[0242]
另外,如上所述并且如本领域中已知的,hel308解旋酶属于sf2家族并且是3'至5'解旋酶(其还可以称为a型解旋酶)。多个解旋酶的核心域可以包括另一种的常见基序,如reca结合折叠含有参与核苷酸结合和水解的walker a基序(其还可以称为基序i)和walker b基序(其还可以称为基序ii)和基序vi。为了解详情,参见flechsig等人,“in silico investigation of conformational motions in super家族2解旋酶蛋白s,”plos one:6(7):e21809(2011)。另外,sf2家族的解旋酶可以共有9个保守基序,其可以称为q、i、ia、ib、ii、iii、iv、v和vi。由于基序ii的序列(dead(seq id no:2)或deah(seq id no:3)或dexh),sf2解旋酶家族还可以称为dead
‑
箱(seq id no:2)蛋白或者deah
‑
箱(seq id no:3)解旋酶。包含在sf2家族中的解旋酶包括recq
‑
样家族和snf2
‑
样家族。多种sf2解旋酶是a型,其
中有些例外,如xpd家族。sf2家族的x
‑
射线晶体衍射研究表明解旋酶保守基序在蛋白三级结构中密切相关,并且它们可以形成大的功能域。为了解详情,参见tuteja等人,“unraveling dna helicases:motif,structure,mechanism and function,”european journal of biochemistry271(10):1849
‑
1863(2004)和hall等人,“helicase motifs:the engine that powers dna unwinding,”molecular microbiology 34:867
‑
877(1999)。由tuteja改编而来的图16示意性示出了已在sf2家族中鉴别的多个基序,例如,dead
‑
箱(seq id no:2)解旋酶,其中hel308是成员。如tuteja所述,开放框(open boxes)代表保守基序。每个解旋酶基序的共有序列由单字母编码表示,例如,图16中“c”可以是d、e、h、k或r;图16中“o”可以是s或t;和图16中“x”可以是任何氨基酸。图16中还示出了分配给基序的名称,例如,q、i、ia、ib、ii、iii、iv、v和vi。如以上进一步提及的,基序i可以称为walker a基序,并且在tuteja中称为atp酶a walker i,基序ii可以称为walker b基序,并且在tuteja中称为atp酶b walker ii。箭头所指的基序之间的数字是插入基序之间的氨基酸残基的典型范围。
[0243]
另外,如wo 2013/057495中所述,hel308解旋酶可以包括氨基酸基序q
‑
x1
‑
x2
‑
g
‑
r
‑
a
‑
g
‑
r(seq id no:66),其中x1可以是c、m或l;x1可以是c;x2可以是任何残基,包括疏水性或中性残基,如a、f、m、c、v、l、i、s、t、p或r。任选地,上述基序中末端r可以与p偶联。
[0244]
考虑到本文所提供的教导和指导,本领域技术人员可以通过确定序列身份或者与以上举例说明的hel308解旋酶中的一个或多个比对来确定参考解旋酶是否是hel308解旋酶。
[0245]
另外,考虑到本文所提供的教导和指导,本领域技术人员可以适合地突变hel308解旋酶以减缓多核苷酸通过孔的部分易位,例如,通过减缓hel308解旋酶实施的水解步骤,通过以可以减缓水解的方式与另一种蛋白质的同源基序类似地突变hel308的基序。作为一个实例,tanaka等人,“atpase/helicase motif mutants of escherichia coli pria protein essential for recombination
‑
dependent dna replication,”genes to cells 8:251
‑
261(2003),描述了在其保守atp酶/dna解旋酶基序(即walker a、b和qxxgrxgr基序)中具有氨基酸取代的pria蛋白(dexh
‑
型解旋酶)的突变体。根据tanaka,某些突变体在某些条件下在水解atp时高度妥协,并且在某些条件下所有的walker a和walker b突变体蛋白显示出高度减弱的dna解旋酶活力。因此,可以预计与tanaka中公开的那些类似的hel308解旋酶的walker a和walker b基序的突变可以预期会减弱dna解旋酶活力或减缓atp水解,可以预期它减缓多核苷酸通过孔的部分易位并因此提高该多核苷酸的表征。作为另一个实例,hishida等人,“role of walker motif a of ruvb protein in promoting branch migration of holliday junctions:walker motif a mutations affect atp binding,atp hydrolyzing,and dna binding activities of ruvb,”journal of biological chemistry 274(36):25335
‑
25342(1999),描述了作为atp
‑
依赖性六聚dna解旋酶的大肠杆菌(escherichia coli)ruvb蛋白的突变体。根据hishida,walker基序a的某些点突变影响ruvb的atp水解和atp结合活性,以及dna结合、六聚体形成和促进分枝迁移的活性。因此,可以预计与hishida中公开的那些类似的hel308解旋酶的walker a基序的突变可以预期会影响atp水解和atp结合,在某些实施方式中,可以预期它减缓多核苷酸通过孔的部分易位并因此可以提高该多核苷酸的表征。
[0246]
因此,本发明公开提供了表征目标多核苷酸的方法。所述方法可以包括:(a)跨越与hel308解旋酶和目标多核苷酸接触的孔施加势差;(b)测量通过所述hel308解旋酶的所述目标多核苷酸通过所述孔的一个或多个分步易位步骤所产生的一个或多个信号;和(c)根据通过所述分步易位步骤产生的所述一个或多个信号,表征所述目标多核苷酸。
[0247]
本发明公开还提供了表征目标多核苷酸的方法,其中所述势差包括势差。还提供了表征目标多核苷酸的方法,其中所述信号包括电信号或光信号。所述电信号可以选自电流、电压、隧穿、电阻、电位、电压、电导率和横向电测量的测量。所述电信号包括通过孔的电流。
[0248]
在其它方面,本发明公开提供了表征目标多核苷酸的方法,其中所述分步易位步骤包括hel308解旋酶完整易位循环的第一分步易位步骤。所述分步易位步骤还可以包括hel308解旋酶完整易位循环的第二分步易位步骤。目标多核苷酸的易位可以处于施加于通过孔易位的多核苷酸上的作用力的相反的方向,或者处于施加于通过孔易位的多核苷酸上的作用力的方向。
[0249]
另外提供了表征目标多核苷酸的方法,其中使用得自完整易位循环的两个分步步骤的电信号表征了目标多核苷酸中的一个或多个核苷酸残基,与使用得自完整易位循环的单一电信号表征一个或多个核苷酸相比,准确度大于50%。
[0250]
还提供了表征目标多核苷酸的方法,其中所述孔为生物孔。所述生物孔可以是多肽孔或多核苷酸孔。在一些方面,多肽孔具有5个核苷酸或以下的缢缩区。在其它方面,多肽孔包括耻垢分枝杆菌(mycobacterium smegmatis)孔蛋白a(mspa)。mspa可以具有seq id no:1所示或与seq id no:1具有至少15%、至少20%、至少25%、至少30%、至少35%、至少40%、至少45%、至少50%、至少55%、至少60%、至少65%或至少70%同源性的氨基酸序列。
[0251]
还提供了表征目标多核苷酸的方法,其中所述孔为固态孔或者生物和固态杂交孔。所述生物和固态杂交孔包括多肽
‑
固态杂交孔或者多核苷酸
‑
固态杂交孔。
[0252]
本发明公开另外提供了表征目标多核苷酸的方法,其中所述hel308解旋酶是表1和2中所示的解旋酶或其变体。还提供了表征目标多核苷酸的方法,其中所述目标多核苷酸选自单链、双链和部分双链多核苷酸。
[0253]
在一些实施方式中,根据所述分步易位步骤所产生的一个或多个信号表征多核苷酸包括应用改进型维特比算法。
[0254]
在一些实施方式中,所述方法还包括步骤(c)之后的(d),改变至少一个参数以改变通过所述hel308解旋酶的所述目标多核苷酸通过所述孔的一个或多个分步易位步骤的时机;和(e)使用改变的至少一个参数,重复步骤(a)
‑
(c)。所述方法还可以包括合并步骤(c)和(e)期间产生的信号并基于所述合并的信号表征所述目标多核苷酸。在一些实施方式中,所述改变的至少一个参数选自温度、盐浓度、辅因子浓度、atp产物(如无机焦磷酸)浓度、adp浓度、ph和所使用的特定hel308解旋酶。
[0255]
在一些实施方式中,表征包括检测和鉴别一种或多种信号中的电平以及基于检测和识别的电平确定和输出目标多核苷酸的序列。
[0256]
说明性地,所述检测和鉴别一种或多种信号中的电平包括输出全电平(full level)、分电平(fractional level)、所有电平和电平标识符(level identifier)中的一
个或多个。
[0257]
基于检测和识别的电平,所述确定和输出目标多核苷酸的序列可以包括将全电平、分电平、所有电平和电平标识符中的一个或多个作为输入,基于所述输入调用多个序列,和基于所调用的多个序列的相关置信信息选择并输出至少一个调用序列。
[0258]
基于检测和鉴别的电平,所述确定和输出目标多核苷酸的序列可以包括将全电平、分电平、所有电平和电平标识符中的一个或多个作为输入,基于所述输入调用多个序列,和基于所调用的多个序列的部分的相关置信信息选择所调用的多个序列的部分并将其彼此连接。
[0259]
基于所述检测和识别的电平,所述确定和输出目标多核苷酸的序列可以包括将全电平、分电平、所有电平和电平标识符中的一个或多个作为输入,基于所述输入调用多个序列,将所调用的序列与模型序列比较,和基于所调用的序列与模型序列比较的相关置信信息选择和输出至少一个调用序列。
[0260]
基于所述检测和识别的电平,所述确定和输出目标多核苷酸的序列可以包括将全电平、分电平、所有电平和电平标识符中的一个或多个作为输入,基于所述输入调用多个序列,将所调用的序列与模型序列比较,和基于所调用的多个序列的部分与模型序列比较的相关置信信息选择所调用的多个序列的部分并将其彼此连接。
[0261]
本发明公开还提供了调节目标多核苷酸通过孔的分步易位步骤的方法。所述方法可以包括:(a)跨越与hel308解旋酶和目标多核苷酸接触的孔施加势差;(b)将所述hel308解旋酶与一定浓度的hel308解旋酶底物接触,所述hel308解旋酶底物的浓度不同于所述底物的参比浓度,所述底物浓度在分步易位步骤持续时间中所产生的变化与所述底物浓度与所述参比浓度相比的差异成正比,和(c)测量目标多核苷酸通过孔的分步易位步骤所产生的信号。步骤(b)可以类似地包括使用底物类似物或抑制剂来实现分步易位步骤持续时间的变化。因此,可以在本发明公开的方法中使用本文所述的或本领域中已知的任何底物类似物或核苷酸抑制剂以用于调节hel308解旋酶底物、用作参比浓度的hel308底物中的任一个或hel308解旋酶底物和用作参比浓度的hel308底物两者的分步易位步骤。
[0262]
能够调节多核苷酸的分步易位步骤的hel308解旋酶底物可以是能够被解旋酶水解的核苷酸或核苷酸类似物。核苷酸底物提供了使双链或部分双链多核苷酸解开或者使单链多核苷酸易位通过孔的能量。hel308解旋酶的常见底物包括(例如)atp。hel308解旋酶底物还包括能够被解旋酶水解的核苷酸和核苷酸类似物。
[0263]
如本文所述,与核苷酸底物结合有关的一个或多个分步易位步骤的停留时间可以与hel308解旋酶底物浓度成反比。例如,在一些测试条件下,每个核苷酸易位所观察到的两个分步易位步骤中仅有一个的停留时间与hel308解旋酶底物浓度成反比。因此,一个易位步骤可以对底物浓度敏感,而另一个易位步骤不敏感。
[0264]
可以通过改变hel308解旋酶底物的浓度来实现获得不同分步易位步骤长度的分步易位步骤的调节。可以确定调节程度或幅度,从而本领域技术人员可以选择适合于所需目标多核苷酸表征的特定分步易位步骤长度。可以通过将hel308解旋酶置于不同于底物参比浓度的hel308解旋酶底物浓度来确定调节程度。与参比浓度相比底物浓度的变化导致了不同的分步易位步骤停留时间,所述停留时间与相比于参比浓度的底物浓度差异成正比。
[0265]
因此,可以通过使用不同于底物参比浓度的hel308解旋酶底物浓度来调节目标多
核苷酸通过孔的分步易位步骤。解旋酶溶液内的其它组分或反应条件也可以用于改变分步易位步骤的停留时间,并因此改变单个易位循环的分步易位步骤长度。不同的分步易位步骤可以类似地用于获得额外的信号信息以提高目标多核苷酸表征的准确度。
[0266]
影响(例如)底物结合至hel308解旋酶的动力学和解旋酶的底物水解的反应组分和反应条件可以用于改变分步易位步骤的停留时间。这些其它因素包括(例如)反应条件的温度、金属浓度,包括二价金属浓度、离子浓度、溶剂粘度。例如,上述因素和条件以及磷酸盐和/或焦磷酸盐浓度可以影响水解步骤。另外,孔两侧的电压可以(例如)影响底物结合和/或构成hel308解旋酶停留时间的解旋酶暂停(解旋酶pause)。其它因素包括(例如)ph、阳离子类型或二阶阳离子浓度和类型、解旋酶突变等,所有都可以影响停留时间。在这点上,例如,提高焦磷酸盐浓度可以用于减缓hel308解旋酶的催化速度并因此提高停留时间。此外,例如,原钒酸钠和腺苷5'
‑
(β,γ
‑
亚氨基)三磷酸锂盐水合物也可以用于减缓解旋酶活性。在以下实施例v中举例说明了调节解旋酶活性的焦磷酸盐和核苷酸类似物的使用。
[0267]
随着顺序步骤之间电流差的提高,使用分步状态进行数据分析的益处也提高。第一次进行近似法时,分步易位步骤将使用相邻完整易位步骤中间的值。如果分步易位步骤比1/2核苷酸(0.3埃)小得多,则在一些情况下或者甚至在大多数情况下,分步值可能难以或甚至不可能观察到。如果分步易位步骤正好是1/2核苷酸长度,那么平均地,所得电流可以最大程度地不同于对应于完整核苷酸步骤的之前和后续电流值。酶的修饰可以允许聚合物亚基进行纳米分数(fractions of a nanometer)的复位。这可以通过提高或降低酶活性水解位点的相对高度至纳米孔的限制缢缩的酶修饰进行。在一些实施方式中,这可以通过添加或移除解旋酶的氨基酸或用具有较大水力学半径的氨基酸的取代来实现。在其它实施方式中,这可以通过改变氨基酸的电荷来实现,所述氨基酸的电荷可以改变对纳米孔边缘的静电斥力或引力。不希望受任何理论束缚,如果“基于紧握”的假设是正确的(如参考图3更详细地描述的),有可能特定突变将影响解旋酶向上推动解旋酶
‑
多核苷酸复合物的程度,其可以转化为核苷酸z轴易位百分比的变化。
[0268]
考虑了精细调节分步易位步骤的持续时间:预期解旋酶atp酶结构域的某些突变将影响atp水解速率是合理的。反过来,这将预期会影响分步易位步骤之一的停留时间。例如,如果水解速率缓慢,那么预期会提高分步易位步骤之一的停留时间。其它突变可以影响atp结合至解旋酶的速率(kon)。在这种情况下,随着atp结合所花费的时间的提高,相应分步易位步骤的停留时间将提高。
[0269]
hel308解旋酶的参比浓度可以是(例如)目标多核苷酸表征中常用的底物的量或者它可以是不同的。例如,如果hel308解旋酶底物的浓度是常用的1.0mm,那么1mm将对应于参比浓度。参比浓度可以经验性地来源于或得自本领域中熟知的报道。在该具体实例中,除1mm之外的底物浓度将是不同于参比浓度的hel308解旋酶底物浓度。如以下进一步描述的,可以使用hel308解旋酶底物的多个浓度和参比浓度以调节或确定分步易位步骤的变化量。
[0270]
可以改变hel308解旋酶底物浓度和参比底物浓度,只要两浓度不是饱和浓度。说明性地,hel308解旋酶底物的饱和浓度为约1mm核苷酸底物。因此,如果参比浓度是1mm,那么要改变的hel308解旋酶底物浓度可以是小于1mm的任何浓度,其包括(例如)0.1μm、1.0μm、10μm、100μm、200μm、300μm、400μm、500μm、600μm、700μm、800μm、900μm。基于hel308解旋酶底物浓度和/或参比,其它示例浓度可以是(例如)1.0mm、2.0mm、3.0mm、4.0mm和4.9mm或更
小。类似地,只要它们不同,hel308解旋酶底物和参比底物浓度两者的浓度可以是未饱和的浓度。因此,hel308解旋酶底物和参比浓度可以是以上所列的任何示例性浓度以及它们之间的任何浓度,例如,0.01μm至5mm的任何浓度范围和该范围之间的所有浓度。
[0271]
相对于表征目标多核苷酸的方法,可以如上所述实施本发明所述的用于调节分步易位步骤的方法。一旦确定了适合于特定需要的hel308解旋酶底物浓度,则该底物浓度可以在本文所述的用于表征目标多核苷酸的方法中使用。以类似方式,可以以影响(例如)底物结合动力学和水解的反应组分和条件实施类似确定,以确定适合于特定需要的组分浓度或反应条件。然后,可以在本发明所述的用于表征目标多核苷酸的方法中使用该适合的浓度或条件。新的底物浓度、反应组分浓度和/或反应条件将导致不同的停留时间,其可以提供附件信号信息,从而以参考实施例ix的如下所述的方式用于提高测定的准确度。
[0272]
因此,本发明公开提供了调节目标多核苷酸分步易位步骤的方法,其进一步包括根据一个或多个分步易位步骤的一个或多个信号,表征目标多核苷酸。表征可以包括鉴别以下中的一个或多个:(1)目标多核苷酸的序列;(2)目标多核苷酸的修饰;(3)目标多核苷酸的长度;(4)目标多核苷酸的身份;(5)目标多核苷酸的来源;或(6)目标多核苷酸的二级结构。
[0273]
本发明公开还提供了调节目标多核苷酸分步易位步骤的方法,其中所述方法使用了势差,所述势差包括势差。还提供了调节目标多核苷酸分步易位步骤的方法,其中分步易位步骤所产生的信号包括电信号或光信号。另外,提供了调节目标多核苷酸分步易位步骤的方法,所述电信号是选自电流、电压、隧穿、电阻、电位、电压、电导率和横向电测量的测量。所述电信号也可以是通过孔的电流。
[0274]
还提供了调节目标多核苷酸分步易位步骤的方法,其中底物浓度是hel308解旋酶底物的半饱和浓度。在一些实施方式中,参比浓度是hel308解旋酶底物的饱和浓度。在其它方面,底物浓度和参比浓度两者是hel308解旋酶底物的半饱和浓度。还提供了调节目标多核苷酸分步易位步骤的方法,其中hel308解旋酶底物是三磷腺苷(atp)。
[0275]
还提供了调节目标多核苷酸分步易位步骤的方法,其中所述分步易位步骤包括hel308解旋酶完整易位循环的第一分步易位步骤或hel308解旋酶完整易位循环的第二分步易位步骤。目标多核苷酸的易位可以处于施加于通过孔易位的多核苷酸上的作用力的相反的方向,或者处于施加于通过孔易位的多核苷酸上的作用力的方向。
[0276]
本发明公开还提供了调节目标多核苷酸分步易位步骤的方法,其中使用得自完整易位循环的两个分步步骤的电信号表征目标多核苷酸中的一个或多个核苷酸残基,与使用得自完整易位循环的单个电信号的一个或多个核苷酸的表征相比,其准确度大于50%。在本发明公开所述方法的一些方面,与参比浓度相比,在较低底物浓度时,表征目标多核苷酸中的一个或多个核苷酸残基具有更大的准确度。
[0277]
还提供了调节目标多核苷酸分步易位步骤的方法,其中所述孔是生物孔。所述生物孔可以是多肽孔或多核苷酸孔。在一些方面,多肽孔具有5个核苷酸或以下的缢缩区。在其它方面,多肽孔包括耻垢分枝杆菌(mycobacterium smegmatis)孔蛋白a(mspa)。mspa可以具有seq id no:1所示或与seq id no:1具有至少15%、至少20%、至少25%、至少30%、至少35%、至少40%、至少45%、至少50%、至少55%、至少60%、至少65%、至少70%、至少75%、至少80%、至少85%、至少90%、至少95%或至少99%同源性的氨基酸序列。
mgcl2和10mm hepes/koh缓冲液,ph 8.0的实验缓冲液。将野生型hel 308tga或野生型phi29聚合酶中的任一种用作马达。在存在hel308 tga的情况下,缓冲液添加1mm atp。在存在phi29的情况下,缓冲液添加dctp、datp、dttp和dgtp各100μm。
[0291]
图2a
‑
2c示出了根据一些实施方式的phi29聚合酶和hel308 tga解旋酶易位事件的比较。图2a示出了与使用phi29 dna聚合酶(dnap)所观察的易位步骤相比,使用hel308 tga解旋酶所观察的分步易位步骤。易位多核苷酸(seq id no:67:/5phos/aaaccttccxcccgtaccgtgccgtaccgttccgttccgtaccgta tttttttt tctcactatcgcattctcatgcaggtcgtagcc,其中x=无碱基)杂交至含有胆固醇的多核苷酸(seq id no:68:cctgcatgagaatgcgatagtgaga tttttttttttttttttttt
‑
cholteg)。使用mspa
‑
m2纳米孔。对hel308 tga解旋酶多核苷酸易位所观察到的电平数目几乎是对phi29 dnap所观察到的电平数目的两倍。在示踪之间划线以表明相应电平。phi29示踪(顶部)是共有事件(consensus),而hel308解旋酶示踪(底部)是所测量的单个易位事件。共有事件可以表示来自对相同序列的多次读取的可靠检测电平的组合。这种组合潜在可能比单个读取更可靠,这是因为它可能不必需包括错误,如单个分子易位所发生的错误,例如,核苷酸“跳跃”或者核苷酸“切换”,如本领域中已知的。
[0292]
图2b示出了将使用phi29聚合酶作为分子马达易位通过mspa
‑
m2纳米孔的单链多核苷酸模板所产生的预期电流电平与使用hel308 tga解旋酶作为分子马达所观察到的那些相比,用hel308 tga解旋酶观察到的分步易位步骤。易位多核苷酸(seq id no:69:/5phos/ctcacctatccttccactxxccccctttgggtttaaattttttcagatctcactatc tttttaaagttttctcactatcgcattctcatgcaggtcgtagcc,其中x=无碱基)杂交至含有胆固醇的多核苷酸(seq id no:68:cctgcatgagaatgcgatagtgagatttttttttttttttttttt
‑
cholteg)。phi29示踪(顶部)是预测的,而hel308解旋酶示踪(底部)是所测量的单个易位事件。如共有事件一样,预测可能不必需包括错误,如单个分子易位可能发生的错误,例如,核苷酸“跳跃”或者核苷酸“切换”,如本领域中已知的。预测可以表示基于k聚体表的模拟数据,该表是先前采集的。phi29预测图形基于每个碱基的完整步骤,并且示出了可以使用完整步骤分子马达预期的一类图形。比较地,明显可以看出hel308 tga解旋酶具有分步步骤。
[0293]
图2c示出了与使用phi29 dnap所观察的易位步骤相比,使用hel308tga解旋酶所观察的分步易位步骤。易位多核苷酸序列是:seq id no:70:/5phos/catcatcatcatcatcatxxccccctaaacaagaataccacgactagcatttttcagatctcactatcgcattctcatgcaggtcgtagcc。所述易位多核苷酸杂交至含有胆固醇的多核苷酸(seq id no:68:cctgcatgagaatgcgatagtgagatttttttttttttttttttt
‑
cholteg)并通过mspa
‑
m2纳米孔。使用简单重复序列5'
‑
cat
‑
3'显示重复图形。对hel308 tga解旋酶多核苷酸易位所观察到的电平数目是对phi29 dnap所观察到的电平数目的两倍。phi29和hel308解旋酶示踪两者均是共有示踪。共有示踪的使用可以有利于不同分子马达之间易位步骤尺寸的比较,并且可以降低或消除潜在可以使解释复杂化的假象,如跳跃和切换。
[0294]
不希望受任何理论束缚,在分步易位步骤的进一步解释中,提出了“基于紧握”的机制。图3示出了根据一些实施方式对分步易位步骤所提议的“基于紧握”的机制。多核苷酸(黑色实线)与解旋酶(水平线填充的图形)结合。一旦atp结合(步骤1),解旋酶经历构象变化(步骤2)。由于解旋酶握紧多核苷酸,因此多核苷酸相对于解旋酶的位置不必需改变。解
旋酶上的参考点(灰色三角形)相对于解旋酶握紧的多核苷酸(参见握紧的多核苷酸上的参考点,灰色正方形)不移动。解旋酶构象变化将解旋酶
‑
多核苷酸复合物从纳米孔顶部推出,同时也将多核苷酸拉入孔缢缩中(带有箭头的黑线所指出的黑线)。第二多核苷酸参考点(白色圆圈)显示构象变化期间(步骤2)多核苷酸相对于孔缢缩移动,其导致分步步骤所测量的电流改变。最后,atp水解并且解旋酶沿多核苷酸易位(步骤3)。这导致多核苷酸相对于解旋酶和孔移动完整核苷酸。总之,在第一分步易位步骤中,hel308解旋酶结合至atp并经历构象变化,该构象变化拉动解旋酶握紧的多核苷酸并通过一个核苷酸的一部分使多核苷酸移距,这反过来产生了可测量的电流变化。在第二分步易位步骤中,atp水解并且hel308解旋酶完成了一个核苷酸通过纳米孔的易位。可以适合地使用其它机制来解释本发明的分步易位步骤的观察结果。
[0295]
实施例ii
[0296]
atp浓度和分步易位步骤之间的关系
[0297]
实施例ii描述了atp浓度对分步易位步骤停留时间的影响。
[0298]
为了进一步阐明分步易位步骤的生物化学机制,在不同的atp浓度下检验分步易位步骤的停留时间。首先用由400mm kcl、10mm hepes,ph 8组成的缓冲溶液填充顺式和反式孔。通过在~25μm直径teflon孔上涂覆十六烷和脂肪的混合物形成由dphpc组成的脂质双分子层,并进行电导测定以确保脂质双分子层和teflon孔之间的千兆欧姆密封(gigaohm seal)。使用连接到一对ag/agcl电极(该电极连接至顺式和反式孔)的axopatch 200b膜片钳放大器进行所有电学测量。膜形成后,将mspa纳米孔注入顺式孔中,其中通过电导测定监控纳米孔向脂质双分子层的掺入。一旦将单个纳米孔掺入到双分子层,灌注顺式室以防止多个孔插入。然后,以10nm的最终浓度将单链多核苷酸注入顺式室,对膜施加电压,并通过瞬间电流响应检测多核苷酸通过孔的易位。多核苷酸易位检测时,将电压设置为0v,并且将1mm mgcl2、115nm hel308解旋酶和各个浓度的atp(10μm、30μm、100μm和1mm)注入到顺式孔内。然后,将电压设置为保持电位(0.01、0.1和1mm atp为140mv;0.03mm atp为180mv),记录电流。如图14a
‑
14d所示,在注入顺式孔内之前,易位多核苷酸(seq id no:71:/5phos/catcatcatcatcatcatxxccccctaaacaagaataccacgactagcatttttcagatctcactatcgcattctcatgcaggtcgtagcc,其中x=无碱基)杂交至含有胆固醇的多核苷酸(seq id no:68:cctgcatgagaatgcgatagtgagatttttttttttttttttttt
‑
cholteg)(如在本文其它处更详细地描述的)。以这种方式,多核苷酸的5'端首先易位通过纳米孔,然后通过hel308解旋酶的加工,再拉回通过纳米孔。axopatch放大器以50khz的采样率并且以10khz的低通滤波器记录系统的电流响应。在该频率范围内,由于多核苷酸的hel308解旋酶加工通过纳米孔的步骤转换(包括分步易位步骤)是明显可鉴别的。实验后,使用计算机算法鉴别多核苷酸易位事件。通过使用确定邻近值之间统计学显著性的本领域熟知的学生t检验,鉴别这些易位事件内统计学显著的电流电平(有关进一步详细情况,参见carter等人,在本文其它处引用,或john e.freund,mathematical statistics,第5版,prentice hall)。对于从该特定序列所观察的电流,与单一步骤分子马达相比,由于存在加工的核苷酸,因此所鉴别的统计学显著的电流电平几乎是两倍,其中拓扑结构(电流电平的峰值和谷值)在每个峰值之间和每个谷值之间具有几乎两倍的水平,如通过直接观察所测量的。
[0299]
为了减少实验误差,在核苷酸易位的大分辨率区域中进行多核苷酸通过纳米孔易
位的持续时间的数据分析。在链测序中,并且具体地,在多核苷酸的纳米孔测序中,由于当与相邻多核苷酸序列相比时,离子流中的显著性差异阻断,因此无碱基区域可以获得相对高的信噪比。出于该原因,与一些不明确的“噪音”影响相比,无碱基区域的附近区域内的统计学显著性水平潜在可以更可能是由于通过纳米孔的核苷酸加工所造成的。出于该原因,选择了围绕并包含无碱基电流峰值的27个电流电平的持续时间来进行基于电流电平持续时间的数据分析。
[0300]
图4a和图4b示出了根据一些实施方式atp浓度对分步易位步骤停留时间的示例性影响。在图4a中,将来自包含分步易位步骤的易位通过纳米孔的多核苷酸序列的无碱基电流峰值的电流电平顺序标记为1
‑
27,并对中值持续时间作图。
[0301]
通过使用学生t检验的算法检测电流电平以确定相邻电流值之间的统计学显著性(参见carter等人,在本文其它处引用)。其它技术能够进行该检测,包括速度阈值和卡方(chi
‑
squared)最小化,并且在本领域中这些对于核苷酸加工
‑
相关电流变化以及对于图像处理中的步骤检测均是已知的。存在与这些电平中每一个相关的持续时间,并且在与穿越孔的相同序列的多个多核苷酸的相同电平的比较中,计算每个电平的中值持续时间。因此,这些中值持续时间代表了与每个电平相关的典型持续时间。然而,由于停留时间的指数分布,这些停留时间的时间常数更指示了它们的atp依赖性。出于该原因,在图4b中,用指数衰减曲线(a*e
‑
t/τ
)对偶数和奇数电平(其中“偶数”和“奇数”与图4a中的电平指数有关)的持续时间柱状图拟合,并对每一个的时间常数作图。通过将每个多核苷酸易位事件中的每个电平的持续时间引入到相同大小的容器(equivalently
‑
sized bins)中,构建电平持续时间的柱状图。然后,使用商用曲线拟合算法(the matlab curve fitting toolbox)对这些柱状图拟合,所述算法使用最小二乘法将数据与指数衰减模型拟合。该方法将残差平方和最小化,其中残差定义为数据点和对该点的拟合反应之间的差异。这是将数据与参数模型拟合的标准技术。图4b中的误差线对应于每次拟合的95%置信界限(confidence bounds)。
[0302]
如图4b所示,偶数电平的停留时间随atp浓度降低而升高,而奇数电平的停留时间保持恒定。因此,偶数电平的停留时间对应于第一分步易位步骤,其表面上与atp结合有关并且与atp浓度成反比,并呈现指数分布,而奇数电平的停留时间对应于第二分步易位步骤,其表面上与atp水解有关并且是atp依赖性的。
[0303]
实施例iii
[0304]
多核苷酸测序中分步易位步骤的使用
[0305]
实施例iii描述了与使用得自完整易位循环的单个电信号相比,通过使用得自完整易位循环的两个分步易位步骤的电信号测序准确度提高。
[0306]
由于mspa“读取
‑
头(read
‑
head)”对缢缩区内的4个核苷酸(4
‑
聚体)延伸敏感,因此从测量对应于mspa纳米孔中所见的所有4
‑
聚体组合的电流的四聚体图中产生了电流示踪。有关测量对应于4
‑
聚体组合的电流的进一步详细信息,参见laszlo等人,“decoding long nanopore sequencing reads of natural dna,”nature biotechnology 32:829
‑
833(2014)。然而,应理解不同的孔可以对缢缩区内不同数目的核苷酸敏感。在本实施例中,如下所述通过比较使用原始de bruijn序列的隐马尔可夫模型(hmm)结果,确定了序列准确度。对于典型的实验噪声水平(如图5中所示,即~0.5
‑
2pa,或约0.5至1.5pa),与分步步骤(正方形)相比,使用完整步骤(菱形)的重建准确度降低。
[0307]
简要地,使用先前所述的方法建立孔(参见butler等人,proc.natl.acad.sci.usa,105:20647
–
20652(2008);manrao等人,plos one,6:e25723(2011))。简要地,从1,2
‑
二植烷酰基
‑
sn
‑
甘油基
‑3‑
胆碱磷酸(avanti极性类脂)在teflon中形成跨过~20微米直径水平孔的脂质双分子层。双分子层两侧上的隔室含有10mm hepes,ph 8.0、400mm kcl、1mm dtt和10mm mgcl2的实验缓冲液。将axopatch
‑
200b(axon instruments)用于在双分子层两侧施加电压(140mv或180mv)并测量离子电流。以~2.5ng/ml的浓度,将mspa加入至底部顺式隔室(cis compartment)。一旦将单个mspa蛋白插入teflon孔,则顺式隔室内冲入实验缓冲液以抑制或避免进一步插入。所有实验在23℃进行。使用4
‑
极贝塞耳滤波器以20khz对模拟离子电流信号低通滤波并使用national instruments 6363数字化仪以100khz数字化。使用以labwindows/cvi写入的常规软件(national instruments)控制数据采集。使用matlab写入的常规软件(the mathworks)分析数据。除了atp滴定实验外,通常以1mm使用atp,在这种情况下,atp浓度范围在10um至1mm。以10nm使用杂交至含有胆固醇的多核苷酸的易位多核苷酸。以115nm的最终浓度使用hel308 tga解旋酶。将多核苷酸和atp加入至顺式室,最后加入hel308 tga解旋酶。作为另外一种选择,可以使用在本领域中熟知的atp再生系统。一个示例性系统包括2mm atp、10mm磷酸肌酸二钠盐、3.5u/ml肌酸激酶和0.6u/ml无机焦磷酸酶。
[0308]
图5根据一些实施方式,对完全步骤(菱形)和步骤(正方形)绘制了电流迹线电子产生的序列重构准确度(隐马尔可夫模型(hmm))对不同水平的附加噪声的图。图5示出了来源于对于de bruijn序列(256
‑
聚体)的模型电流阻断示踪的hmm/viterbi算法分析的序列重建准确度。在一些方面,一般hmm算法类似于timp等人,biophys j.2012may 16;102(10):l37
‑
9.doi:10.1016/j.bpj.2012.04.009中所述的算法。该算法可以从一系列观察测量恢复潜在的m“状态”组。该算法的基本形式依赖于两个通过实验确定的概率组:状态
‑
状态“转移”概率和状态
‑
观察“发出”概率。对于n个测量,在步骤i=1、2、3
……
n中给出测量。一个概率组是转换矩阵,其描述了对于给定时间i和状态s
i
(其中s是m状态组中的状态),后续状态s
i+1
(其中s
i+1
不必需是s
i
)的概率。对于纳米孔系统,通过对4nt敏感并且检验4个规范核苷酸(a、c、g、t)的纳米孔,导致44=256个状态,其对应于4nt的每个组合。这些状态中的每一个仅可以转换为4个相邻状态之一。
[0309]
图6a示出了根据一些实施方式hmm解码纳米孔中序列所需的具有非零概率的状态转移,其中多核苷酸是通过马达酶移动的。所述马达为在1个核苷酸的步骤中移动多核苷酸的phi29 dnap或类似的酶。图6b示出了根据一些实施方式hmm解码纳米孔中序列所需的具有非零概率的状态转移,其中多核苷酸是通过马达酶移动的。所述马达为使聚合物能够分步移动的hel308解旋酶或类似的酶。
[0310]
在图6a中对于在单个核苷酸步骤中移动的酶示出了该系统转换矩阵的非零转换概率。使用这类酶,每个多核苷酸状态或n聚体必需达到4个相邻n
‑
聚体状态之一。对于采取一个分步易位步骤的酶,将有更多状态。在这点上,在可以观察到另一种完整步骤状态之前,给定完整步骤状态必须进入半步(或分步步骤)状态。因此,通过更多可辩别的途径,存在更多可用的状态,因此辅助了多核苷酸表征的准确度。
[0311]
通过q*4
n+1
提供了状态数,其中n是纳米孔的读取大小,而q是完成完整易位循环所需的步骤数。对于q=2和n=4,如对hel308解旋酶和m2
‑
nnn mspa所观察的,存在2048个状
态。在图6b中图示描述了在分步核苷酸步骤中移动的酶的转移概率矩阵。对应于完整状态的每个状态仅可以转移至1024个“半状态”或“分步状态”之一,而每个半(或分步)状态可以转移至4个不同的状态,其对应于纳米孔读取头内的新的状态。对于hmm解码算法,使用另一个概率组:在时间t的电流测定c
t
属于状态s
i
的概率。通过实验确定或根据先前实验观察估计概率组。可以通过比对算法的迭代应用来完成这种估计,如laszlo等人2014(在本文其它处引用)中所述或者通过hmm的期望最大化来完成。为了评价分步易位步骤的使用,将使用分步核苷酸步骤的酶的测序准确度与使用单个核苷酸步骤的酶的相比较。通过在matlab中进行的常规软件实施hmmviterbi译码算法,并对每个条件产生10个蒙特卡洛模拟电子实验。根据这100个蒙特卡洛模拟的平均和标准偏差获得了测序平均值和标准偏差。基于manrao 2012(在本文其它处引用)的结果,产生电流电平。以图5中x
‑
轴所示的值给出的高斯宽度,加入高斯噪声以改变在序列重构中使用的电子观察电流值。,并且典型的纳米孔测序实验具有约1parms的平均水平波动。对于所添加的0.5pa宽度的高斯偏移,分步和完整步骤重构两者均获得了100%的测序准确度测量。在具有高于0.5pa的宽度的添加的高斯偏移之上,分步易位步骤的测序准确度大于非分步易位步骤的测序准确度。因此,当将大于0.5pa的高斯噪声加入至平均电流电平时,分步易位步骤的其它信息提供或给予了提高的序列重构准确度。
[0312]
除噪声波动之外,由酶的随机运动所造成的跳跃水平将或可以预期会降低测序准确度。相邻四聚体中核苷酸图形的重读会或可以部分补偿这种准确度的降低。通过添加分步易位步骤,存在核苷酸图形的其它重读。例如,在相邻分步步骤中包含了给定k聚体的相关信息,因此在那些相邻分步步骤期间重读了k聚体。例如,假设具有序列atcgtc的多核苷酸正分布易位通过具有4
‑
核苷酸敏感的读取头的纳米孔。不希望受任何理论束缚,对于完整步进马达,仅当c和g之间的区域位于读取头中心时读取4
‑
聚体tcgt(也就是说,先前“步骤”将使“tc”位于读取头中心,并且仅读取atcg;下一步将使
‘
gt’位于读取头中心并仅读取cgtc)。因此,如果马达跳过tcgt读取步骤,则不再测量与特定4
‑
聚体有关的信息。然而,不希望受任何理论束缚,通过分步步进马达,如hel308解旋酶,在完整步骤期间,两个相邻核苷酸之间的区域可以位于读取头中心,而在分步步骤期间,单个核苷酸可以位于读取头中心。因此当上述多核苷酸的
‘
cg’位于读取头中心时,读取tcgt,如在完整步进情况中一样。先前分步步骤可以仅具有位于读取头中心的c,并且可以读取atcgt相关信息;后一分步步骤仅具有位于读取头中心的g,并读取tcgtc相关信息。由于在分步步骤情况中,
‘
tcgt’相关信息可以读取3次,而在完整步进情况下,仅读取1次,因此4
‑
聚体的这种额外的“重读”可以使得能够获得tcgt相关信息,所述马达跳过了与之相关的步骤—这在完整步进情况中可能是错误的。对于移除水平的所有部分,测序准确度存在2
‑
8%的改善。这通过实施电流电平随机去除的额外的电子蒙特卡罗模拟进行显示。总之,对于纳米孔测序实验中所见的错误形式,测序准确度存在稳健提高。图10、11和12示出了方案,通过该方案,额外的分步易位步骤信息可以用于改善序列准确度。在以下实施例vi中还举例说明了这些方案。所述方案是示例性使用,并且不意欲限制。
[0313]
实施例iv
[0314]
分步易位步骤在图形匹配中的使用
[0315]
实施例iv描述了使用已知算法的分步易位步骤的示例性使用以识别电平。使用动
态规划算法,如needleman
‑
wunsch比对,额外的水平为在多个水平内准确找到图形提供了帮助。有关needleman wunsche比对算法的进一步详细信息,参见durbin等人,biological sequence analysis,第11版(cambridge university press,cambridge,uk 2006)。除电平电流平均值之外或作为它的替代,使用电平持续时间、电平电流标准偏差或电平分布可以进一步提高图形匹配准确度。在本实施例中,使用needleman wunsch比对算法来鉴别对应于包含在对应于1000个碱基的序列的电平内的15个碱基的序列的电平。比较了下列使用:(1)对应于完整核苷酸移动的电平、(2)对应于2个半(或分步)步骤移动的电平、(3)对应于2个半步骤(或分步步骤)移动的电平和持续时间。通过从不同宽度的高斯分布中产生的随机值所提供的值迁移的电平,通过10个蒙特卡罗模拟电子产生观察结果。图7示出了结果,其示出了根据一些实施方式发现作为高斯偏移的函数的电流图形的预期准确度。根据10个蒙特卡罗模拟的平均和标准偏差产生比对准确度的平均和标准偏差。图7中,菱形示出了具有完整核苷酸步骤的马达,而圆圈示出了具有分步易位步骤的马达,正方形示出了具有分步易位步骤结合持续时间值的马达。简要地,将对应于15个核苷酸的电平图形包含在对应于随机1000个核苷酸的序列的电平图形内。电平对应于具有完整核苷酸步骤(菱形)的马达,如phi29 dnap(仅完整易位步骤)或对应于具有分步易位步骤的马达(圆形),如hel308解旋酶。除了电流值之外,使用持续时间以进一步改善匹配(正方形)。根据图7中的结果,可以理解对于提高的噪音,对于使用分步易位步骤移动的算法,匹配质量显著更好。如果还使用持续时间值,则将进一步改善匹配质量。为了将电平与needleman wunsch算法相匹配,使用输入水平相似性测量或得分比较水平。在测试中,使用学生t检验比较电流电平。为了比较两个持续时间的相似性(或对其打分),确定了持续时间的自然对数差异,并将其加入到学生t检验给出的得分中。可以在needleman wunsch算法的术语表中定义术语“得分”。这些打分功能代表了可以用于比较信号电平(例如,电流值)和持续时间的方法的非限制性实例。
[0316]
实施例v
[0317]
调节解旋酶分步步骤
[0318]
实施例v举例说明了改变反应组分对改变hel308解旋酶停留时间的使用。
[0319]
图8示出了根据一些实施方式随焦磷酸盐浓度的改变,hel308解旋酶活性的示例性调节。图9示出了根据一些实施方式用核苷酸抑制剂原钒酸钠和用核苷酸类似物腺苷5'
‑
(β,γ
‑
亚氨基)三磷酸锂盐水合物对hel308解旋酶活性的示例性调节。
[0320]
通过提高焦磷酸盐浓度调节hel308解旋酶活性。简要地,反应条件为实施例iii中所述的那些。通过包含0至50mm范围内的多个浓度的焦磷酸盐,例如,0mm(对照)、5mm、10mm、20mm、30mm、40mm和50mm。图8示出了结果,并且示出了与在不存在焦磷酸盐(对照)的情况下的解旋酶活性相比的解旋酶活性的百分比。5和10mm焦磷酸盐浓度导致解旋酶活性降低至对照的大于75%。大于10mm的焦磷酸盐浓度导致解旋酶活性进一步降低,并因此降低了解旋酶的停留时间。使用荧光测定监控解旋酶打开双螺旋dna的能力。49
‑
nt fret多核苷酸(50nm最终浓度)包含5'荧光素基团(/fam/)。含有40
‑
nt猝灭剂的多核苷酸(50nm最终浓度)包含荧光猝灭剂black hole quencher(/bhq1/)。使用本领域熟知的方法,通过将它们加热超过它们的解链温度至75℃并缓慢冷却至室温将这两条多核苷酸杂交在一起。双螺旋包括3'至5'解旋酶可以结合的9
‑
碱基3'突出。与含有40
‑
nt猝灭剂的多核苷酸100%互补的互补fret 40
‑
nt多核苷酸以至少10倍摩尔过量存在。由于起初猝灭剂和荧光团非常接近,因此
荧光淬灭。基于打开双螺旋dna的解旋酶,含有40
‑
nt猝灭剂的多核苷酸与再结合至49
‑
nt fret多核苷酸相比,更可能结合至互补fret 40
‑
nt多核苷酸。因此,在存在适合的激发光源的情况下,新的单链49
‑
nt fret多核苷酸发荧光。测定缓冲液包含10mm hepes,ph 8.0、400mm kcl、1mm mgcl2、1mm dtt、1mm atp。读取荧光前,使反应在室温下进行20分钟。
[0321]
在分别存在核苷酸抑制剂或类似物原钒酸钠和腺苷5'
‑
(β,γ
‑
亚氨基)三磷酸锂盐水合物的情况下,hel308解旋酶活性并且因此停留时间也显示降低。简要地,反应条件是实施例iii中所述的那些。在5mm抑制剂或类似物浓度下,包含原钒酸钠(图9中的“原钒酸钠”)或腺苷5'
‑
(β,γ
‑
亚氨基)三磷酸锂盐水合物(图9中的“amp
‑
pnp”)。图9示出了结果,并且示出了与在不存在核苷酸抑制剂或类似物(对照)的情况下的解旋酶活性相比的解旋酶活性的百分比。5mm抑制剂或类似物浓度导致解旋酶活性降低至大于对照的85%,并因此可以预期会提高解旋酶停留时间或解旋酶沿dna移动所花的时间。例如,停留时间增加可以延长分步步骤的时间,因此允许更长的信号采集时间。
[0322]
实施例vi
[0323]
处理分步步骤信息以改善测序准确度的方法
[0324]
实施例vi举例说明了处理得自分步易位步骤的额外信息以改善测序准确度的三种方法。
[0325]
图10示出了使用电流电平和持续时间信息处理得自分步易位步骤的额外信息的示例性方法。所述方法可以应用于两个独立序列读取。使用该方案,对电流示踪进行步骤检测算法,其中发现了电流电平和那些电平的持续时间。至少部分基于所述电平的持续时间,两状态hmm将电平鉴别为完整步骤(长)或者半(或分步)步骤(短)或者观察结果内的电位跳跃。然后,通过hmm、viterbi或图形匹配算法或其适当组合使用这些鉴别的长和短步骤以及跳跃信息来分别对两个状态类型重建多核苷酸序列(长和短分别对应于完整步骤和半(或分步)步骤电平)。然后,比较调用序列并用于改善多核苷酸测序准确度,例如,通过调节hmm、viterbi或图形匹配算法。比对可以用于较差地鉴别两个独立序列读取的匹配位置。
[0326]
图11示出了使用电流电平和持续时间信息处理得自分步易位步骤的额外信息的示例性方法。所述方法可以应用于两个并行的序列读取。在该方法中,首先对电流示踪进行步骤检测算法以发现电平。然后,将平均(或中值)电平电流值以及每个电平的持续时间成对输入到检验持续时间和电流值并对半(或分步)状态以及完整状态估计或调用最优序列的二维hmm、viterbi或图形匹配算法或其适合的组合中。在该技术中,hmm发射概率不是二维的:p
i
(发射
t
)=p
i
(cur
t
,dur
t
)=p
i
(cur
t
)*p
i
(dur
t
),其中i是对应于多核苷酸的长步骤或短(分步)易位步骤的“状态”,cur
t
和dur
t
分别是电平数t的电平电流和持续时间。二维hmm可以作为输入采取长电平(完整状态)的共有图谱和概率分布以及短电平(半(或分步)状态)的共有图谱和概率分布。二维hmm可以作为输出提供核苷酸序列的调用。
[0327]
图12示出了直接使用电流示踪处理得自分步易位步骤的额外信息的示例性方法。可以通过或不通过使用持续时间信息应用所述方法。通过参考持续时间信息的使用,在该方法中,使用持续时间
‑
依赖性hmm直接分析电流示踪。在这种hmm形式中,同时将电平持续时间确定为最可能的序列和完整或者半(或分步)步骤状态。如果在两个时间迭代之间状态保持不变,则将提高给定状态的持续时间。然后,该持续时间用于改善状态是处于完整状态还是分步状态的评价。
[0328]
实施例vii
[0329]
处理分步步骤信息以改善测序准确度的其它方法
[0330]
实施例vii描述了处理分步步骤信息以改善测序准确度的其它示例性方法
[0331]
基于来自使用单步分子马达易位通过纳米孔的多核苷酸的信号,先前已将隐马尔可夫模型(hmm)和viterbi算法用于碱基调用(base
‑
calling)。为了解详情,参见timp等人,“dna base
‑
calling from a nanopore using a viterbi algorithm,”biophysical journal 102:l37
‑
l39(may 2012)。图19a示意性示出了来自用于表征多核苷酸通过孔的单步易位的信号的示例性隐马尔可夫模型(hmm)的方面,例如,其中给定信号电平对应于一个核苷酸通过孔的易位,例如,通过聚合酶或解旋酶通过孔的易位。如本文在其它处所提及的,信号电平可以不必需对应于孔缢缩内单个核苷酸的存在,但是可以对应于包含多个核苷酸,例如,2、3、4、5、6、7、8、9、10或10个以上的核苷酸的“单词”的存在。这个“单词”也可以称为“k
‑
聚体”。在图19a所示的实施方式中,所述“单词”或“k
‑
聚体”是4个核苷酸长或者是“四聚体”或“4
‑
聚体”,其对应于基于孔缢缩中4个核苷酸存在的信号电平。
[0332]
在图19a中,可以看出对于易位通过孔的多核苷酸的给定位置i,孔缢缩中给定的四聚体可以包括四种核苷酸的任何可能组合,例如,aaaa、aaac、aaag、aaat
……
tttt。基于对应于该四聚体的信号电平来唯一地鉴别该四聚体可以不必是可能的。例如,预期序列中两个不同的四聚体,例如,彼此临近的两个不同的四聚体潜在地可以具有与另一个相同的信号电平。timp公开了dna三联体(3
‑
聚体)的示例性电流值,基于此可以看出某些三联体可以具有与另一个相同的信号电平,因此抑制了碱基的核苷酸碱基调用,其中所述三联体仅基于对应于该三联体的电流电平。应理解某些四聚体(并且,更一般地,某些k
‑
聚体)可以具有彼此不可区分的信号电平,因此抑制了碱基的核苷酸碱基调用,其中四聚体或k
‑
聚体仅基于对应于该4
‑
聚体或k
‑
聚体的电流电平。因此,使用hmm的术语,基于信号电平的观察,彼此不可区分的这类四聚体或k
‑
聚体中的碱基可以作为“隐状态”建模。
[0333]
可以使用基于孔缢缩中多核苷酸的其它单步位置的观察结果的其它信息,从而提高准确鉴别该四聚体或k
‑
聚体中的碱基,并因此准确鉴别“隐状态”的可能性。例如,在图19a中,还可以看出对于易位通过孔的多核苷酸的下一位置i+1,孔缢缩中给定四聚体可以仅具有4个核苷酸的某些可能组合,这是因为位置i的最后三个核苷酸对应于位置i+1的前3个核苷酸。照此,对于i和i+1状态的信号测量可以用于提高正确鉴别在i和i+1位置(或者,同样地,i
‑
1和i位置)中的一个或两个处存在的四聚体的可能性。例如,基于对应于多核苷酸的位置i的序列aaaa,对于位置i+1,仅四个序列aaaa、aaac、aaag和aaat是可用的。对于位置i的每个可能序列,可以容易地鉴别位置i+1的可用的4个序列。类似地,基于多核苷酸位置i+1处的序列,可以容易地鉴别多核苷酸位置i+2处可用的4个序列。用于单步马达的viterbi算法——其中信号电平和位置i、i+1、i+2、
……
、i+n之间存在一对一的对应,其中n是多核苷酸中核苷酸的数目,可以表示来自有序电平组l={l1,l2,
…
l
n
}的信号。对应于多核苷酸位置i的每个电平l
i
可以表示为该信号电平的平均值(平均值
i
)、该信号电平的标准偏差(std
i
)或该信号电平的持续时间(dur
i
)中的一个或多个。当对应于多核苷酸现有位置(i位置)的四聚体是q时,可能的四聚体组可以表示为定义对应于多核苷酸前一位置(i
‑
1位置)的四聚体的可能值的prev(q)={q1,q2,
…
q4}。例如,prev(aacc)={aaac,caac,gaac,taac}。基于对应于jth观察电平的观察信号电平o
ji
,位置i处存在的给定四聚体q的可能性
得分可以表示为:
[0334][0335]
其中s(l
i
|q)对应于表示观察水平l
i
,给定的四聚体q的可能性的奖励得分,inspen是插入罚分(罚分对应于所观察到的但不对应于多核苷酸中四聚体的信号电平),delpen是缺失罚分(罚分对应于多核苷酸中不具有对应信号电平的四聚体)。
[0336]
图19b示意性示出了根据一些实施方式用于表征来自使用hel308解旋酶的多核苷酸通过孔的分步易位的信号的示例性hmm的方面。在图19b中,还可以看出对于易位通过孔的多核苷酸的给定位置i,孔缢缩中给定的四聚体可以包括四种核苷酸的任何可能组合,例如,aaaa、aaac、aaag、aaat
……
tttt。可以使用基于孔缢缩中多核苷酸的分步步骤位置以及其他单步位置的观察结果的其它信息,从而以相对于仅使用单独的单步位置的改善的准确度,提高准确鉴别该四聚体或k
‑
聚体中的碱基,并因此准确鉴别“隐状态”的可能性。
[0337]
例如,在图19b中,还可以看出对于分步步骤马达,易位通过孔的多核苷酸的下一位置是“i分步”并且其中孔缢缩中给定四聚体可以仅具有4个核苷酸的某些可能组合,这是因为位置i的最后三个核苷酸对应于位置“i分步”的前3个核苷酸。照此,i和i分步状态的信号测量可以用于提高正确鉴别所提供的四聚体的可能性。例如,基于对应于多核苷酸位置i的序列aaaa,对于位置i分步,仅4个序列aaaa、aaac、aaag和aaat是可用的。对于位置i处每个可能的序列,可以容易地鉴别位置i分步处可用的4个序列。
[0338]
另外,在图19b中,还可以看出对于易位通过孔的多核苷酸的下一位置i+1完整(该位置紧挨着i分步),孔缢缩中给定四聚体可以仅具有1个可能序列,这是因为位置i+1完整的4个核苷酸对应于位置i分步的相同核苷酸。照此,对应于i、i分步和i+1完整位置的信号测量可以用于提高正确鉴别i、i分步和i+1完整位置(或者,同样地,i
‑
1和i位置)中一些或全部处存在的四聚体的可能性。例如,基于对应于多核苷酸位置i的序列aaaa,对于位置i分步和对于i+1完整,仅4个序列aaaa、aaac、aaag和aaat是可用的。用于分步步骤马达的改进型viterbi算法
–
其中信号电平和分步步骤以及完整步骤位置i、i分步、i+1完整、i+1分步、i+2完整、i+2分步、
……
、i+n分步、i+n完整之间存在对应关系,其中n是多核苷酸中核苷酸的数目,可以作为电平组表示信号电平l。类似地,如以上参考图19a所讨论的,对应于i完整或i分步位置的每个信号电平l
i
可以表示为该信号电平的平均值(平均值
i
)、该信号电平的标准偏差(std
i
)或该信号电平的持续时间(dur
i
)中的一个或多个。考虑到四聚体q,在电流分步易位步骤中,对应于先前完整位移步骤的可能四聚体组可以定义为prev(q)={q1,q2,
…
q4}。例如,prev(aacc)={aaac,caac,gaac,taac}。
[0339]
基于对应于位置i的观察信号电平l
i
,对于对应于完整易位状态的位置,位置i处存在的给定四聚体q的可能性得分score
f
以及对于对应于半(或分步)易位状态的位置,位置i处存在的给定四聚体q的可能性得分score
h
可以如下表示为:
[0340]
[0341][0342]
其中s
f
(l
i
|q)对应于表示观察电平l
i
,完整易位状态的给定四聚体q的可能性的奖励得分,s
h
(l
i
|p,q)对应于表示观察电平l
i
,分步易位状态的给定四聚体q和前一四聚体p的可能性的奖励得分,inspen是插入罚分(罚分对应于所观察到的但不对应于多核苷酸中四聚体的信号电平),delpen是缺失罚分(罚分对应于多核苷酸中不具有对应信号电平的四聚体)。
[0343]
另外,动态规划可以用于分步步骤分子马达(如hel308解旋酶)的图形匹配。在laszlo等人,“decoding long nanopore sequencing reads of natural dna,”nature biotechnology 32:829
‑
833(2014)中对于单步分子马达描述了动态图形匹配。例如,对于单步分子马达,信号电平l可以表示为电平组l={l1,l2,
…
l
n
},其中对应于多核苷酸的完整易位步骤位置的每个信号电平l
i
可以表示为该信号电平的平均值(平均值
i
)、该信号电平的标准偏差(std
i
)或该信号电平的持续时间(dur
i
)中的一个或多个。基于观察的信号电平l
i
,测量的给定四聚体q
j
的可能性得分可以表示为:
[0344][0345]
其中i代表电平序列中的位置;j代表dna序列中的位置,四聚体q
j
的最后一个碱基将是位置j处的碱基;得分(i,j)代表电平l1…
l
i
和四聚体q1…
q
j
之间匹配的良好程度;s(l
i
|q
j
)对应于表示观察电平l
i
,给定四聚体q
j
的可能性的奖励得分;inspen是插入罚分(罚分对应于所观察到的但不对应于多核苷酸中四聚体的信号电平);delpen是缺失罚分(罚分对应于多核苷酸中不具有对应信号电平的四聚体)。
[0346]
对于分步步骤分子马达(如hel308),信号电平l可以表示为电平组l={l1,l2,
…
l
n
},其中对应于多核苷酸的i完整或i分步位置的每个信号电平l
i
可以表示为该信号电平的平均值(平均值
i
)、该信号电平的标准偏差(std
i
)或该信号电平的持续时间(dur
i
)中的一个或多个。基于观察的信号电平l
i
,对应于完整易位状态的测量的给定四聚体q
j
的可能性得分score
f
以及对应于半(或分步)易位状态的测量的给定四聚体q
j
的可能性得分score
h
可以如下表示为:
[0347][0348][0349]
其中i代表电平序列中的位置;j代表dna序列中的位置,四聚体q
j
的最后一个碱基将是位置j处的碱基;score
f
(i,j)和score
h
(i,j)代表分别假定为完整或分步状态的电平
l1…
l
i
和四聚体q1…
q
j
之间的匹配良好程度;s
f
(l
i
|q
j
)和s
h
(l
i
|q
j
)分别对应于表示观察电平l
i
,处于完整或分步状态的给定四聚体的可能性的奖励得分;inspen是插入罚分(罚分对应于所观察到的但不对应于多核苷酸中四聚体的信号电平);delpen是缺失罚分(罚分对应于多核苷酸中不具有对应信号电平的四聚体)。
[0350]
现将参考图20a描述使用分步步骤的一些示例性从头测序结果。基于人dna产生了75个500
‑
聚体多核苷酸文库,并且如在本文其它地方所述的参考实施例ii和iii类似地采集纳米孔数据。使用以上方程(2)和(3),使用所述的改进型viterbi算法,分析基于数据的核苷酸碱基调用。然后,将碱基调用序列与150个500
‑
聚体组比对,其中75个是正确的500
‑
聚体,并且其中75个是“诱饵”或“虚假”500
‑
聚体序列。在图20a中,该图示出了作为比对准确度函数的读取长度(使用lastal aligner,如kielbasa等人,“adaptive seeds tame genomic sequence comparison,”genome research21:487
‑
493(2011)中所述),空心菱形对应于其中碱基调用序列与正确(“目标”)序列比对的结果,而实心菱形对应于其中碱基调用序列与“诱饵”或“虚假”序列比对的结果。根据图20a可以理解对于大于约200个基底对的读取长度,可以获得大于约60%的准确度。还可以使用已知的技术,如读取dna的两条链来潜在地提高准确度。
[0351]
现将参考图20b
‑
20c描述使用分步步骤的一些示例性图形匹配结果。如上所述,参考图20a使用75个500
‑
聚体多核苷酸的相同文库和相同实验规程。使用以上方程(5)和(6),使用所述的用于图形匹配的动态规化,分析基于数据的核苷酸碱基调用。然后,将碱基调用序列与150个500
‑
聚体组比对,其中75个是正确的500
‑
聚体,并且其中75个是“诱饵”或“虚假”500
‑
聚体序列。在图20b中,该图示出了作为比对得分的函数的比对尺寸,空心菱形对应于其中碱基调用序列与正确(“目标”)序列比对的结果,而实心菱形对应于其中碱基调用序列与“诱饵”或“虚假”序列比对的结果。根据图20b可以理解对于大于约200个碱基对的比对尺寸可以获得大于约40的比对得分。在图20c中,该图也示出了作为比对得分的函数的比对尺寸,空心菱形对应于其中碱基调用序列与正确(“目标”)序列比对的结果,而实心菱形对应于其中碱基调用序列与“诱饵”或“虚假”序列比对的结果。根据图20c可以理解对于大于约50个碱基对的比对尺寸可以获得大于约20的比对得分。可以观察到分步步骤模型可以比单步模型准确鉴别到更多事件。
[0352]
另外,可以看出对于使用1332个电平的易位事件,针对80kb数据组的图像匹配(方程5和6)对单链花费了145秒,而使用1332个电平的相同事件,针对该数据组的从头测序(方程2和3)对单链花费了69秒。可以看出图形匹配复杂性随核苷酸数据组线性增加,而从头序列的复杂性独立于数据组。观察图形匹配以准确鉴别较短的事件,这是从头测序不能鉴别的。另外,可以看出图形匹配的分步步骤模型比单步模型产生了更多真阳性结果,这表明分步步骤模型可以是更好的模型以解释解旋酶数据。
[0353]
实施例viii
[0354]
使用其它hel308解旋酶的分步易位步骤
[0355]
实施例viii描述了使用示例性hel308解旋酶作为分子马达所观察到的分步易位步骤。
[0356]
如上所述,使用dphpc脂质双分子层中的单个2nnn mspa纳米孔并且使用下表3中所列的参数,参考实施例i类似地实施实施例viii的实验,其中“hel308 mbu(a)”是指用于
使用hel308 mbu的第一实验的一组参数,并且“hel308 mbu(b)”是指用于使用hel308 mbu的第二实验的一组参数。脂质双分子层由1,2
‑
二植烷酰基
‑
sn
‑
甘油基
‑3‑
胆碱磷酸(avanti极性脂)形成。双分子层在teflon中横跨水平直径~20微米的孔。以~2.5ng/ml的浓度,将m2
‑
nnn
‑
mspa加入至双分子层的底侧(grounded side)。一旦插入单个孔,则隔室内冲入实验缓冲液以避免进一步插入。axopatch
‑
200b膜片钳放大器(axon instruments)将180mv的电压施加到双分子层两侧并测量离子电流。使用4
‑
极贝塞耳滤波器以50khz低通滤波模拟信号,然后以低通滤波频率的5倍数字化。使用以labwindows/cvi写入的常规软件(national instruments)控制数据采集。双分子层两侧~60μl的隔室含有适当浓度的kcl、1mm edta、1mm dtt、1mm atp、5mm mgcl2和10mm hepes/koh缓冲液,ph 8.0的实验缓冲液。以对分子马达指明的浓度使用野生型mbu hel 308解旋酶。
[0357]
在hel308 mbu实验和hel308 tga实验中,以3'至5'的方向读取dna,而在phi29聚合酶实验中,以5'至3'的方向读取dna。
[0358]
表3
[0359][0360]
图17a
‑
17d示出了根据一些实施方式使用某些参数的hel308 mbu解旋酶、hel308 tga解旋酶和phi29聚合酶易位事件的比较。图17a示出了使用表3中所示的“hel308 mbu(a)”参数,通过hel308 mbu解旋酶观察到的易位步骤。易位多核苷酸(seq id no:72:/5phos/aaaccttccxcccgtaccgtgccgtaccgttccgttccgtaccgtatttttttttctcactatcgcattctcatgcaggtcgtagcc,其中x=无碱基)杂交至含有胆固醇的多核苷酸(seq id no:73:aaaaaaaatacggtacggaacggaacggtacggcacggtacgggtttttttttttttttt/3cholteg)。图17b示出了使用表3中所示的“hel308 mbu(b)”参数和使用图17a中相同的多核苷酸序列通过hel308mbu解旋酶所观察到的易位步骤。图17c示出了使用表3中所示的“hel308tga”参数和使用图17a中相同的多核苷酸序列通过hel308 tga解旋酶所观察到的易位步骤。图17d示出了使用表3中所示的“phi29”参数和使用图17a中相同的多核苷酸序列通过phi29聚合酶所观察到的易位步骤;对纵轴反映图17d中的phi29图,以有利于图17a、17b、17c和17d之间的比较。
[0361]
从图17a
‑
17d可以看出,对于使用每种解旋酶的测序,纳米孔检测了通常表示为“a”(对应于信号谷值)和“b”(对应于信号峰值)的特征。还可以看出对于使用hel308 tga解旋酶的测序(图17c),相比于phi29解旋酶(图17d),观察到了约两倍的电平数。还可以看出对于在“hel308mbu(b)”条件下使用hel308 mbu解旋酶的测序(图17b),相比于在“hel308mbu(a)”条件下使用hel308 mbu解旋酶的测序(图17a),观察到了更多的电平数。还
可以看出对于在“hel308 mbu(b)”条件下使用hel308 mbu解旋酶的测序(图17b),相比于hel308 tga解旋酶(图17c),观察到了较少的电平,而相比于phi29解旋酶(图17d),观察到了较多的电平。图17a
‑
17d可以理解为表示(1)hel308解旋酶(例如,tga和mbu两者)的多个变体示出了分步步骤,而对于聚合酶phi29未观察到分步步骤;和(2)通过改变环境变量或参数,例如,kcl浓度,可以阐明分步步骤。另外,其它数据表明当使用mbu时,电平持续时间随atp浓度的降低而增加,例如,mbu中的持续时间以及因此所造成的分布步骤的物理机制也可以是atp
‑
依赖性的。
[0362]
实施例ix
[0363]
应激物的使用,任选地与多种方式结合
[0364]
基于本文所提供的发明公开应清楚多种环境变量或参数可以影响纳米孔系统如何读取或如何基于特定多核苷酸序列产生信号。可以提供这种影响的示例性变量或参数可以包括温度、盐浓度(例如,mg、cl)、辅因子(例如,atp)浓度、atp产物如焦磷酸盐的浓度、ph、所使用的具体的分子马达(例如,所使用的具体的hel308解旋酶)、压力等。
[0365]
例如,如上所述参考实施例ii和图4a和4b,atp的浓度可以影响对应于某些易位步骤的电平的停留时间。例如,观察到第一分步易位步骤的停留时间随atp浓度的降低的增加,并且表面上与atp结合有关并与atp浓度成反比。作为另一实例,如上所述参考实施例v和图8,焦磷酸盐的浓度可以影响hel308解旋酶的活性。例如,观察到hel308解旋酶的活性随焦磷酸盐浓度的升高而降低,因此提高了解旋酶的停留时间。作为另一实例,如上所述参考实施例v和图9,核苷酸抑制剂或类似物的浓度可以影响hel309解旋酶的活性。例如,观察到基于原钒酸钠或腺苷5'
‑
(β,γ
‑
亚氨基)
‑
三磷酸锂盐水合物(amp
‑
pnp)的存在,hel308解旋酶活性降低,因此提高了解旋酶的停留时间。作为另一实例,如上所述参考实施例viii和图17a和17b,盐浓度可以影响所观察到的电平的数目。例如,在使用hel308 mbu解旋酶测序期间,观察到盐(例如,kcl)浓度升高提高了所观察到的电平的数目。本领域技术人员将能够容易地设想调整任何适合的参数以调整基于多核苷酸序列产生信号的方式。
[0366]
另外,应理解这些参数的不同组合可以影响测序准确度以及测序通量。例如,解旋酶停留时间的提高可以提高准确度,例如,可以提高所观察到的电平的数目,但是潜在地可以降低测序通量。对于基于分步步骤观察的测序,与另一组步骤相比,一些步骤潜在地可以受特定变量影响更大。变量
‑
独立步骤可以用于设置准确度基线,同时可以调整其它步骤以满足特定测序需要(例如,提高准确度而降低通量,或者提高通量而降低准确度)。在一些实施方式中,多方式装置可以通过基于测序仪的需要来调整准确度和通量而进行利用,例如,通过在测序期间调整一个或多个参数。作为一个非限制性、说明性实例并且如上所述,已观察到使用hel308 tga,atp浓度的降低可以提高分步状态的持续时间。分步状态持续时间的增加可以提高测序准确度,例如,通过改善分步状态读取的信噪比(snr)或允许应用低频滤波器,但是它可以降低通量。多方式装置可以通过以高浓度atp开始测序,从而相对快地确定序列的大致“结构(scaffold)”,然后可以降低atp浓度以通过更高质量,尽管更慢的读取来“填充结构中的空隙”。
[0367]
另外,注意可以顺序或彼此平行地使用任何适合个数的不同参数以提高通过hel308解旋酶的目标多核苷酸通过孔的易位所产生的一个或多个信号的分辨率。图21a
‑
21c示意性示出了根据一些实施方式对于多核苷酸通过孔的不同易位,可以作为时间函数
产生的信号。图21a
‑
21c分别示出了虚线曲线,其对应于其中在仅有势差所施加的外加力的情况下,而不是通过分子马达,多核苷酸易位通过孔的情况下所产生的理想化信号,并且其具有无穷大的信号分辨率。在这些条件下,当它们穿过孔时,信号是核苷酸位置和序列的连续改变函数。
[0368]
图21a也示出了仅使用在垂直虚线所指明的时间发生的完整易位步骤所产生的示例性信号(粗实线)。所述信号可以是电信号或光信号,如本文其它处所述。另外,所述信号可以包括这种电信号或光信号的任何适合的特征,如平均信号电平、信号持续时间或标准偏差(例如,宽带噪声或限带噪声)。在图21a中可以看出通过单个步骤信号从相对低电平改变为相对高电平,然后再通过单个步骤改变为相对低电平,其对应于多核苷酸易位通过孔的完整易位步骤间发生的转换。在图21a中还可以看出信号在不同时间在点(a)、(b)和(c)处与理想化信号相交,并因此可以认为在这些点对理想化信号“采样”。然而,由于有效采样率相对较低,因此所述信号对理想化信号的采样相对较差。例如,点(a)和(b)处的值彼此是相同的,其对应于不同易位步骤的简并信号电平。由于信号没有对点(a)和(b)之间存在的理想化曲线部分充分采样,因此对应于点(a)和(b)的物理易位步骤可以是彼此不可区分的,从而导致多核苷酸序列相关信息的损失。另外,由于信号没有对点(a)和(b)之间存在的理想化曲线部分充分采样,因此对应于点(b)和(c)之间理想化曲线的下降斜率的物理易位步骤潜在地仅可以部分表征这些步骤期间易位通过孔的多核苷酸部分。
[0369]
除了如上所述由虚线曲线表示的理想化信号之外,图21b还示出了使用在垂直虚线所指明的时间发生的时间分隔的完整易位步骤的组合或完整和分步易位步骤的组合所产生的示例性信号(粗实线)。时间分隔的完整易位步骤可以对应于分别易位多核苷酸,但在彼此相对改变(例如,彼此相对改变了完整易位循环持续时间的约50%)的时间的两个分子马达所产生的信号。完整和分步易位步骤的组合可以对应于通过部分和完整易位步骤分步易位多核苷酸的单个分子马达(例如,hel308解旋酶)所产生的信号,如其中所述分步易位步骤在完整易位循环持续时间的约50%发生。所述信号可以参考图21a如上所述。在图21b中可以看出通过步骤序列信号从相对低电平改变为相对高电平,然后再通过另一步骤序列改变为相对低电平,其对应于多核苷酸易位通过孔时的时间分隔的完整易位步骤之间或通过完整和分步易位步骤的组合所发生的转换。在图21b中还可以看出信号在与图21a相比显著更多的点(和时间)与理想化信号相交,并因此可以认为在这些点对理想化信号“采样”。由于比图21a中的有效采样率相对更高,因此所述信号对理想化信号的采样比图21a中相对更好。例如,点(a)和(b)处的值彼此是相同的,其对应于不同易位步骤的简并信号电平。由于图21a中的信号也对点(a)和(b)之间存在的理想化曲线部分采样,因此对应于点(a)和(b)的物理易位步骤可以彼此区分,从而导致产生了多核苷酸序列的相关附加信息。另外,由于在图21b中信号对点(a)和(b)之间存在的理想化曲线部分更充分地采样,因此对应于点(b)和(c)之间理想化曲线的下降斜率的物理易位步骤可以比使用图21a中的信号可以潜在地实现的更好地表征这些步骤期间易位通过孔的多核苷酸部分。
[0370]
除了如上所述由虚线曲线表示的理想化信号之外,图21c还示出了使用在垂直虚线所指明的时间发生的时间分隔的完整易位步骤的组合或完整和分步易位步骤的组合所产生的另一种示例性信号(粗实线)。时间分隔的完整易位步骤可以对应于分别易位多核苷酸,但在彼此相对改变(例如,彼此相对改变了完整易位循环持续时间的约25%、50%和
75%)的时间的多个分子马达所产生的信号。完整和分步易位步骤的组合可以对应于通过部分和完整易位步骤分步易位多核苷酸的单个分子马达(例如,hel308解旋酶)所产生的信号,如其中所述分步易位步骤在完整易位循环持续时间的约25%、50%和75%发生。所述信号可以参考图21a如上所述。在图21c中可以看出通过比图21b中更多个数的步骤序列信号从相对低电平改变为相对高电平,然后再通过比图21b中更多个数的步骤另一序列改变为相对低电平,其对应于多核苷酸易位通过孔时的时间分隔的完整易位步骤之间或通过完整和分步易位步骤的组合所发生的转换。在图21c中还可以看出信号在与图21b相比显著更多的点(和时间)与理想化信号相交,并因此可以认为在这些点对理想化信号“采样”。由于比图21b中的有效采样率相对更高,因此所述信号对理想化信号的采样比图21b中相对更好,并因此可以比使用图21a或21b中的信号潜在可以实现的更好地表征这些步骤期间易位通过孔的多核苷酸。
[0371]
应理解可以使用任何适合的参数选择以提高理想化样品曲线的任何选择部分的采样。例如,如上所述,可以使用来自不同分子马达的时间
‑
改变(相
‑
改变)的完整易位步骤的组合。在这点上,尽管图21b描述了分子马达彼此之间时间改变了完整易位步骤时间的50%,并且尽管图21b描述了分子马达彼此之间时间改变了完整易位步骤时间的25%、50%和75%,但是这些值是纯说明性的,并且分子马达可以彼此之间时间
‑
改变任何适合的时间量,例如,可以彼此之间改变完整易位步骤时间的5%至95%中的任一个量,例如,可以彼此之间改变完整易位步骤时间的10%至90%中的任一个量,例如,可以彼此之间改变完整易位步骤时间的25%至75%中的任一个量,例如,可以彼此之间改变完整易位步骤时间的40%至60%中的任一个量。作为另一实例,完整和分步易位步骤的组合可以对应于通过部分和完整易位步骤分步易位多核苷酸的单个分子马达(例如,hel308解旋酶)所产生的信号。尽管图21b描述了彼此之间在完整易位步骤时间的50%发生的分步易位步骤,并且尽管图21c描述了彼此之间在完整易位步骤时间的25%、50%和75%发生的分步易位步骤,但是这些值是纯说明性的,并且分步易位步骤可以在相对于完整易位步骤的任何适合的时间发生,例如,在完整易位步骤时间的5%至95%,例如,在完整易位步骤时间的10%至90%,例如,在完整易位步骤时间的25%至75%,例如,在完整易位步骤时间的40%至60%。
[0372]
另外,应理解可以通过改变任何适合的参数来适合地调整完整或分步步骤发生的相对时间和因此信号对理想化信号采样的时间。例如,如上所述。可以影响信号产生的示例性变量或参数可以包括温度、盐浓度(例如,mg、cl)、辅因子(例如,atp)浓度、atp产物如焦磷酸盐的浓度、ph、所使用的具体的分子马达等。在一些实施方式中,可以基于第一参数组产生第一信号,从而在第一离散时间组对理想化信号采样,并且可以基于第二参数组(其在至少一个方面不同于第一参数组)产生第二信号,从而在第二离散时间组对理想化信号采样。可以合并第一和第二信号以提供通过比单独的第一或第二信号更大的分辨率对理想化信号采样的信号曲线。应理解可以以类似的方式合并任何适合个数的信号以提供通过比那些信号中任何单独一个更大的分辨率对理想化信号采样的信号曲线。
[0373]
实施例x
[0374]
用于序列鉴别的其它方法
[0375]
参考实施例x描述了用于序列鉴别的一些其它方法。
[0376]
在一些实施方式中,可以单独或彼此结合地使用某些类型的信息以获得序列特异
性信息:(a)单独的完整步骤响应信息、(b)单独的分步步骤响应信息、(c)不包括鉴别器的完整步骤和分步步骤响应信息,和(d)与鉴别器一起的完整步骤和分步步骤响应信息。
[0377]“响应信息”表示得自系统对给定多核苷酸序列(k聚体)的数据,它对k聚体或k聚体的亚组(包括所关心的k聚体在内)是唯一的。响应信息的实例包括平均电流电平、中值电流电平、电流噪声的带宽电平、电流噪声的带限电平、电平持续时间等。
[0378]“鉴别器”表示多核苷酸(k聚体)与纳米孔环境相互作用所获得的数据,其中根据“理想响应”,特定电平相对于其它电平存在。例如,在存在相对高或相对低电平atp浓度的情况下使用hel308 tga解旋酶作为分子马达的系统可以分别对于每隔一个电平显示出相对短或者相对长的持续时间,其中每隔一个电平为根据来自相邻电平的理想响应的约50%。在本实施例中,电平持续时间可以用作鉴别因素,这是因为它可以用于鉴别沿理想响应的序列位置(相对于相邻层)。
[0379]“理想响应”表示系统对通过纳米孔的特定多核苷酸易位的响应,其具有足够高的分辨率从而可以分辨多核苷酸足够小的运动。例如,理想响应是易位通过纳米孔的dna的无限高的分辨率的连续电流示踪。
[0380]
再次提及以上在本实施例中进一步提到的项目(a)
‑
(d),可以独立使用项目(a)
‑
(d)中的每一个或与项目(a)
‑
(d)中一个或多个其它项目结合使用以鉴别多核苷酸序列。例如,由于(例如)计算资源限制、时间限制、优化方法的先验知识等,可以根据项目(a)
‑
(d)中的任何其它项目独立计算项目(a)
‑
(d)中的一个或多个。基于要计算的项目(a)
‑
(d)中不止一个项目,可以仅使用项目(a)
‑
(d)中的一个项目的结果。对于要使用该类计算中哪一个的确定可以基于结果的可信程度。例如,结果的可信性可以基于以下中的一种或多种:(a)响应信息本身(例如,高电平的atp可以缩短hel308 tga中的分布步骤长度,其可以降低项目(b)相对于项目(a)的可信性);(b)测序算法本身(例如,viterbi算法可以对它所提出的最佳序列产生可能性得分,其可以用于确定所提出的序列的可信性水平);(c)测序算法所产生的序列(例如,可以根据算法所提出的序列与序列对照表和/或要测序的多核苷酸的任何先验知识之间的比较来分配可信性);或者(d)项目(a)
‑
(c)的任何适合的组合。
[0381]
注意在一些情况下,通过使用根据项目(a)
‑
(d)中不止一个项目所提出的序列来确定真实序列可以是有益的。例如,可以基于一些或所有这些所提出的序列确定共有序列。可以基于所有或一些所提出的序列来确定共有序列。可以将共有序列全局应用于整个多核苷酸序列或者局部应用于序列的一部分。可以基于来自一些或所有项目(a)
‑
(d)的可信性值来确定共有序列。可信性值可以是以上在本实施例中进一步描述的那些。可信性值可以局部应用于序列的一部分,或者全局应用于整个序列。可以通过多轮上述方法来确定最终的共有序列,其中可以将每轮所获得的共有序列用作所提议的序列,并且每轮之间用于确定每轮的可信性的方法可以是不同的。
[0382]
作为一个实例,viterbi算法可以用于通过仅对易位通过纳米孔的dna的完整步骤或仅对分步步骤测序来确定两个不同的所提议的序列(项目(a)和(b),如本实施例中上述的)。将该算法对每个dna片段的可能性得分用于确定所提议的序列的每个区域的置信度,并且每个区域的置信度的集合体可以导致产生第一轮提议的共有序列。然后,可以就已知序列的对照表将该共有序列与两个初始提议的序列相比较。对照表和这三个提议的序列之间的相似性可以导致对三个提议序列中每个序列的每个区域产生置信值。这三个提议的序
列之间的第二轮基于置信度的比较可以导致产生最终提议的共有序列。
[0383]
在一些实施方式中,图22a
‑
22d示出了根据一些实施方式使用多核苷酸通过孔的分步易位所提供的信息的说明性方法中的步骤。图22a示出了根据一些实施方式使用多核苷酸通过孔的分步易位所提供的信息的方法的高电平概述。图22a中所示的方法包括获得信号(步骤2210),如通过hel308解旋酶的目标多核苷酸通过孔的一个或多个分步易位步骤所产生的一个或多个信号,如本文其它处更详细地描述的。图22a中所示的方法还包括电平检测和识别(步骤2220),例如,检测和鉴别信号中不同的信号电平,例如,检测和鉴别对应于多核苷酸通过孔的分步易位步骤的电平,以及检测和鉴别对应于多核苷酸通过孔的完整易位步骤的电平。图22a中所示的方法还包括序列确定(步骤2230),例如,基于检测和鉴别信号中不同的信号电平来表征多核苷酸的序列。图22a所示的方法还包括输出序列(步骤2240),例如,基于序列调用结果输出实际核苷酸的可能核苷酸序列。
[0384]
图22b
‑
22d示出了图22a中所示的一个或多个步骤的任选的子步骤。例如,图22b示出了图22a所示的步骤2210和2220的一种可能实施的额外细节。图22b中所示的方法还包括获得信号(步骤2210),如通过hel308解旋酶的目标多核苷酸通过孔的一个或多个分步易位步骤所产生的一个或多个信号,如本文其它处更详细地描述的。图22b所示的方法还可以任选地包括获得输入参数(2211)。这些输入参数可以包括(但不限于)定义应检测和确定以对应于信号的那些特征信号的特征的参数。例如,输入参数可以定义信号值中的阈值幅度变化,可以将高于该信号幅度变化检测为对应于电平。或者,例如,输入参数可以限定应检测仅对应于完整易位步骤的信号电平,或仅对应于分步易位步骤的信号电平,或者对应于完整和分步易位步骤两者的信号电平。输入参数还可以含有与错误形式(例如,核苷酸跳跃或核苷酸切换)有关的信息,其可能包括某些错误形式的倾向和/或程度,当确定电平时,可以对其加以考虑。输入参数还可以包括与特定环境有关的信息,在此之间纳米孔、分子马达和多核苷酸相互作用(例如,温度、盐度、ph、辅因子浓度等),这可以用于确定给定信号的电平。图22b中所示的方法还包括电平检测,例如,检测对应于多核苷酸通过孔的分步易位步骤的信号中不同的信号电平(步骤2221)。例如,基于步骤2210获得的信号和步骤2211获得的输入参数,该电平检测可以检测充分统计学显著不同于其它信号区域的信号区域以对应于电平。电平检测的示例性方法(其还可以称为边缘检测或步骤检测)在本领域中是已知的,并且包括学生t检验和卡方最大化。对于可以适合地调整以用于在步骤2221的检测电平中使用的步骤检测算法的一些实例,参见carter等人,“a comparison of step
‑
detection methods:how well can you do?,”biophysical journal 94:306
‑
308(january 2008)。
[0385]
图22b中所示的方法还包括基于步骤2221的电平检测的输出电平信息(步骤2222)。电平信息可以包括对于给定电平检测的平均、中值、形式、分步、持续时间、最大和/或最小电流,或者这些值的任意组合,或者属于给定电平的电流值的亚组的这些值(例如,在首先除去与错误形式有关的电流信息后,可以使用平均电流)。电平信息还可以包括电流的标准偏差或电流的频带限制亚组(例如,在使用低通、高通、带通或带阻滤波器或这些滤波器的任何组合后,获得电流)。电平信息还可以包括与电平持续时间有关的信息以及与电平有关的错误形式信息。图22b中所示方法还包括电平识别(步骤2223),例如,确定在步骤2222输出的电平信息的在步骤2221检测的哪个电平对应于目标多核苷酸的完整或分步易位步骤。例如,步骤2223可以包括分析在步骤2222输出电平信息的在步骤2221检测的不同
电平的持续时间,并且基于这些持续时间,鉴别对应于完整易位步骤的某些电平,并且鉴别对应于分步易位步骤的其它某些电平。作为一个实例,可以认为具有比第一阈值更短的持续时间的信号电平对应于噪音并因此弃去,而可以认为具有比第一阈值更长但比第二阈值更短的持续时间的信号电平对应于分步易位步骤并因此鉴别为该信号,而可以认为具有比第二阈值更长但比第三阈值更短的持续时间的信号电平对应于完整易位步骤并因此鉴别为该信号,而可以认为具有比第三阈值更长的持续时间的信号电平对应于错误或不存在多核苷酸并且因此弃去。
[0386]
图22b中所示的方法还包括输出以下输出中的一种或多种:全电平、分电平、全部电平和电平识别符(level identifiers)。例如,如上所提及的,在步骤2211获得的输入参数可以限定应检测仅对应于完整易位步骤的信号电平,或仅对应于分步易位步骤的信号电平,或者对应于完整和分步易位步骤两者的信号电平(例如,“全部电平”)。注意,在一些实施方式中,通过输入参数选择“全部电平”可以对应于绕过电平识别步骤,从而电平检测步骤2221直接输出全部电平。作为另外一种选择,基于电平检测结果2223和输入参数2211,可以输出所期望的信号的鉴别电平,例如,用于进一步加工,如以下参考图22c和22d所述的。电平识别符可以包括有利于电平进一步分析的任何适合的信息,例如,表明步骤2223期间使用的完整或分步步骤的持续时间以表示所识别的电平对应的转换类型的指数。
[0387]
再参考图22a,可以使用图22b中所示方法或者使用另一种适合的方法产生的全电平、分电平、全部电平和电平鉴别符中的一个或多个可以用作进行序列确定的输入(图22a中的步骤2230)。例如,图22c示出了基于这些全电平、分电平、全部电平和电平识别符中的一个或多个,例如,作为输入的全电平、分电平、全部电平和电平识别符中的一个或多个进行序列确定的第一示例性方法。图22c中所示的方法包括基于全电平、分电平、全部电平和电平识别符中的一个或多个的输入的序列调用步骤(步骤2231)。序列调用可以包括基于输入信号电平的基于目标多核苷酸的哪个核苷酸碱基可以调用的任何适合的方法。序列调用的示例性方法包括(但不限于)viterbi算法,如实施例vii中参考图19a所述,改进型viterbi算法,如实施例vii中参考图19b所述,或类似于实施例xi中所述的图形匹配。可以适合地使用序列调用的其它方法。序列调用的输出(步骤2231)可以包括多种调用序列,例如,序列a、序列b、
…
序列n,以及对于每个该调用序列的置信度信息。不同的调用序列可以基于彼此不同的对步骤2231的输入。例如,第一调用序列(例如,序列a)可以基于对步骤2231的输入,其中基于在步骤2210获得的给定信号,仅鉴别完整易位电平,第二调用序列(例如,序列b)可以基于对步骤2231的输入,其中仅鉴别分步易位电平,和第三调用序列(例如,序列n)可以基于对步骤2231的输入,其中鉴别所有易位电平(例如,完整和分步易位电平两者)。作为另外一种选择或另外,其它调用序列可以基于其它电平,所述其它电平是基于在步骤2211获得的替代输入参数鉴别的,如限定应检测和确定哪种特征信号的特征以对应于信号的参数的不同值,如信号值的不同阈值幅度变化,可以将高于此的信号幅度变化检测为对应于电平。每个不同的调用序列可以具有相关置信度信息,例如,代表调用序列对应于目标核苷酸真实序列的可能性的值。
[0388]
在图22c所示的实施方式中,序列选择步骤(步骤2232)可以选择一个或多个调用序列并作为输出提供选择的序列(步骤2240)。作为一个实例,序列选择步骤(步骤2232)可以包括对多个调用序列比较置信度信息,并且可以在步骤2240选择并输出具有高置信度的
调用序列,例如,对应于真实序列的最高可能性。作为另一个实例,给定调用序列的置信度信息可以包括分别代表调用序列的相应部分对应于目标多核苷酸的该部分的真实序列的可能性的多个置信度值。对于调用序列的不同部分(例如,10个碱基对长,或50个碱基对长,或100个碱基对长,或10
‑
100个碱基对长,或10
‑
50个碱基对长,或50
‑
100个碱基对长的部分),序列选择步骤(步骤2232)可以包括不同调用序列在该部分的置信度值的比较,以及选择对该部分具有最高值的调用序列的部分。所选部分可以与对这些部分具有最高值的其它调用序列的所选部分连结或进行比对。
[0389]
图22d示出了可以用于序列确定的替代方法(2230)。图22d中所示的方法可以包括作为输入获得多个调用序列,例如,序列a、序列b、
…
序列n,以及对于每个该调用序列的置信度信息,其可以类似于参考图22c的上述那些。在这点上,尽管未具体说明,但是图22d中所示的方法可以包括序列调用步骤2231:类似于参考图22c的上述那些的接收输入,类似于参考图22c的上述那些的提供输出和与步骤2231类似的操作。作为另外一种选择,图22d中所示的方法可以从任何其它适合来源获得多个调用序列。
[0390]
图22d所示的方法还可以包括获得模型序列(步骤2234)。例如,这些序列可以包括对于一个或多个不同物种,如一个或多个不同的病原体,先前已知的序列。说明性地,可以将模型序列保存在查找表、数据库或保存在永久计算机可读介质上的其它适合的数据结构中。图22d所示的方法还可以包括序列选择步骤(步骤2233)。在图22d所示的实施方式中,序列选择步骤可以基于在步骤2234获得的一个或多个模型序列,选择作为输入接受的一个或多个调用序列,并作为输出提供所提议的序列和新的置信度信息。作为一个实例,序列选择步骤(图22d中的步骤2233)可以包括将多个调用序列中的一个或多个与在步骤2234获得的一个或多个模型序列相比较,并且可以选择和输出所提议的序列,其可以对应于具有最高的新的置信度信息的调用序列,例如,对应于模型序列的最高可能性。输入置信度信息可以对序列(或者该序列内的区域)的可能性至关重要,以匹配模型序列(模型序列内的区域)以确定最可能的序列,并将其作为提议的序列输出。例如,对于与模型序列z最佳比对的输入序列a和与模型序列y最佳比对的输入序列b,基于a和z之间的比对优于b和y之间的比对,所提议的序列可以是模型序列z。然而,在b和y具有最佳比对的情况下,则a比b具有更高置信度值的情况可以使z成为所提议的序列。另外,在另一种情况中,可以比较序列区域,从而使输出的提议序列包含来自a、b、z和y的序列信息。作为另外一种选择,给定调用序列可以包括新的置信度信息,例如,分别代表调用序列的相应部分对于该部分对应于一个或多个模型序列的部分的可能性的多个新的置信度值。对于调用序列的不同部分(例如,10个碱基对长,或50个碱基对长,或100个碱基对长,或10
‑
100个碱基对长,或10
‑
50个碱基对长,或50
‑
100个碱基对长的部分),序列选择步骤(步骤2233)可以包括将不同调用序列在该部分的新的置信度值与模型序列进行比较,和选择对该部分具有最高的新的置信度值的调用序列的部分。所选部分可以与对这些部分具有最高的新的置信度值的其它调用序列的所选部分连结或进行比对。
[0391]
图22d中所示的方法还可以包括基于步骤2233的新的置信度信息输出,确定对于所提议的序列新的置信度信息以及步骤2233的输出是否满足要求(步骤2235)。作为一个实例,步骤2235可以将新的置信度信息(其可以是新的置信度值)与阈值置信度值相比较,在该阈值置信度值或之上,所提议的序列可以确定为与模型充分匹配,而低于阈值置信度值,
所提议的序列可以确定为与模型未充分匹配。新的置信度信息可以包括输入置信度信息的结果,所提议的序列和输入序列之间的关系,所提议的序列和模型序列之间的关系和/或输入序列和模型序列之间的关系。例如,在所提议的序列只不过是输入序列之一的情况下,新的置信度信息可以是输入序列的输入置信度值的加权平均数以及与最佳比对模型序列的比对得分。在其它情况下,如当所提议的序列是输入序列区域的组合时,新的置信度信息可以包括输入可信度值的加权平均值的加权平均值以及所提议序列的区域中(与模型序列)的比对得分。基于在步骤2235确定新的置信度信息满足要求(“是”),步骤2235作为输出提供所提议的序列(步骤2240)。基于在步骤2235确定新的置信度信息不满足要求,步骤2235返回至2233,此时继续序列选择,例如,通过调用序列与模型序列的进一步比较。序列选择算法或模型序列组可以依赖于参数,其可以包括所提议的序列、新的置信度信息、实施序列选择算法的次数和已使用的模型序列中的一个或多个。例如,初始通过序列选择算法可以使用相对少的模型序列(例如,为了通量)。然而,如果输入序列和模型序列之间的比对相对较差,则新的置信度信息可能不满足要求,并因此一旦返回步骤2233,则可以与新的一个或多个模型序列的精炼组进行比较。
[0392]
实施例xi
[0393]
图形识别,任选地用于snp鉴别
[0394]
在一些实施方式中,本文所公开的方法和组合物可以与多路核酸检测、基因分型和扩增方法组合使用。用于多路核酸检测、基因分型和扩增的方法在本领域中是熟知的并且技术人员可以容易地选择并使用上述方法。例如,在一个实施方式中,本文所公开的方法和组合物可以与以下美国专利中所述的多路核酸检测、基因分型和扩增方法组合使用:美国专利no.6,890,741、6,913,884、7,955,794、7582,420和8,288,103和美国专利公开2013
‑
0244882,以上专利作为参考并入本文。
[0395]
在一些实施方式中,可以与本文所公开的方法和组合物组合的用于多路核酸检测、基因分型和扩增的方法包括在固体载体上实施或与固体载体组合实施的方法,所述固体载体如阵列(随机和有序阵列)或珠。例如,在一些方面,可以将要测定的目标多核苷酸,如基因组dna固定至固体载体。可以对这些固定的目标多核苷酸进行在本领域中熟知的多路核酸检测和基因分型方法。可以使用本文所公开的方法表征所得目标多核苷酸。
[0396]
在一些实施方式中,用于表征目标多核苷酸的方法还可以包括产生要测定的目标多核苷酸所必需的步骤。因此,在一些实施方式中,所述方法可以包括以下步骤:(a)提供从3'至5'分别包含第一、第二和第三目标结构域的多个目标核酸序列,其中第一目标结构域包含检测位置、第二目标结构域为至少一个核苷酸;(b)将目标核酸序列与每个目标序列的探针组接触以形成一组第一杂交复合物,每个探针组包括:第一探针,其从5'至3'包含通用引物序列和与目标序列的第一目标结构域基本互补的序列,以及适合于和检测位置碱基配对的探查位置(interrogation position)(例如,在3'端4个末端碱基内),和第二探针,其从5'至3'包含与目标序列的第三目标结构域基本互补的序列和通用引物序列,其中任选地至少一个探针含有位点鉴别序列(例如,标签或条型码);(c)在一定情况下将杂交复合物与延伸酶和dntp接触,其中如果探查位置处的碱基与检测位置的碱基完美互补,则第一探针发生延伸通过第二目标结构域以形成第二杂交复合物;和(d)将延伸的第一探针与第二探针连接以形成扩增模板。在该方法的一些方面,探针组的第一或第二探针可以包括等位基
因鉴别序列(例如,标签或条型码)。
[0397]
在一些实施方式中,用于表征目标多核苷酸的方法还可以包括以下步骤:(a)提供从3'至5'分别包含第一、第二和第三目标结构域的多个目标核酸序列,其中第一目标结构域包含检测位置、第二目标结构域为至少一个核苷酸;(b)将目标核酸序列与探针接触,所述探针分别从5'至3'包含通用引发序列和与目标序列的第一目标结构域基本互补的序列,以及适合于和检测位置碱基配对的探查位置(例如,在3'端4个末端碱基内),其中任选地所述探针含有位点鉴别序列(例如,标签或条型码);(c)在一定情况下将杂交复合物与延伸酶和dntp接触,其中如果探查位置处的碱基与检测位置的碱基完美互补,则探针发生延伸通过第二和第三目标结构域以形成可以用作扩增模板的延伸探针。
[0398]
产生用于在本文所述的方法中测定的目标多核苷酸的方法还可以包括将扩增模板扩增以产生扩增子。在一些方面,基于已引入扩增模板的鉴别序列,包含第一或第二探针的通用引发序列的引物还包含等位基因鉴别序列或位点鉴别序列(例如,标签或条型码)。可以使用本文所公开的方法表征这些扩增子,所述扩增子可以包含位点鉴别序列和等位基因鉴别序列两者。目标序列的表征可以表明样品的基因分型基于位点和等位基因鉴别序列的存在。
[0399]
在一些实施方式中,用于产生扩增子的引物包括不允许延伸酶在扩增期间使用以穿过残基的一个或多个修饰的残基。例如,在一些方面,一个引物包括无碱基位点(无嘌呤/无嘧啶位点)、c3间隔臂亚磷酰胺(int c3间隔臂)、三甘醇间隔臂(int间隔臂9)或18原子的六乙二醇间隔臂(int间隔臂18)以防止延伸酶继续引物延伸。应理解本领域的技术人员可以选择可以实施这种相同功能的其它修饰的残基。一个或多个修饰的残基可以位于等位基因鉴别序列内或等位基因鉴别序列的任一侧,只要对于使用本文所公开的方法的目标多核苷酸的表征产生了足够长度的5'突出。例如,5'突出具有足够长度以允许扩增子的固定化。
[0400]
在一些实施方式中,通过上述方法产生的扩增子还与切口核酸内切酶接触,从而在第二探针序列中或附近产生3'突出。这些切口酶可以是序列特异性的,从而仅切割双链产物中的一条链。多种切口核酸内切酶在本领域中是熟知的并且认识到基于探针和引发序列,技术人员可以容易地选择适合的核酸内切酶。为了在切口核酸内切酶切割后产生3'突出,可以使用本领域中已知的一些方法,包括(例如)扩增子的部分变性,从而从扩增子释放较小部分的切口链,而扩增子的剩余部分仍保持杂交在一起。为了辅助除去扩增子的较小部分,可以加入所述较小部分的反向互补序列以杂交至不希望的链。
[0401]
在一些实施方式中,可以通过在以上方法中所述的第二探针序列中包含一个或多个尿嘧啶残基来产生3'突出,并将扩增子与在尿嘧啶位置处特异地产生单个核苷酸缺口的尿嘧啶特异的酶接触。这类尿嘧啶特异的酶的非限制性实例是uracil
‑
specific excision reagent(user
tm
)enzyme(new england biolabs)。因此,使用熟知的方法,可以容易地将通过所述酶产生的较小交替片段与扩增子变性开。
[0402]
在某些方面,所产生的3'突出具有足够长度以有利于本文所述的解旋酶的结合。因此,在一些方面,3'突出包括至少4个核苷酸长度。在其它方面,3'突出包括4
‑
20个核苷酸长度,或在某些方面,8
‑
16个核苷酸长度,或在其它方面,包括10
‑
16个核苷酸长度。
[0403]
短语“位点鉴别序列”是指已分配或已知连接至目标多核苷酸上特定位点的核酸残基序列(例如,标签或条型码)。目标多核苷酸的位点可以是(例如)基因组上接近于要测
定的等位基因的基因、基因的一部分(例如,外显子或内含子)或非编码区(例如,启动子或增强子)。位点鉴别序列可以是对所关心的目标序列的位点特异的天然存在的序列和/或对所关心的目标序列非天然的合成序列。可以通过标签或条型码的预期信号图案分配位点鉴别序列。
[0404]
短语“等位基因鉴别序列”是指已分配给目标多核苷酸检测位置中特定核酸残基的核酸残基序列(例如,标签或条型码)。等位基因鉴别序列可以表明检测位置中核酸残基(例如,a、t、c或g)的存在。还可以通过标签或条型码的预期信号图案来分配等位基因鉴别序列。
[0405]
在另一个实施方式中,用于表征目标多核苷酸的方法还可以包括图18中所述的步骤。该方法可以包括以下步骤:(a)提供具有不同的所关心的目标核酸序列的样品,其中所述不同的目标核酸序列任选地固定在固体载体上;(b)将样品与每个不同的所关心的目标核酸序列的探针组接触以形成杂交复合物,每个探针组包括:第一探针,其从5'至3'包含第一通用引发序列和与第一目标结构域基本互补并且具有适合于和检测位置碱基配对的探查位置(interrogation position)的序列;和第二探针,其从5'至3'包含与第三目标结构域基本互补的序列和第二通用引物序列,其中至少一个探针含有对所关心的目标序列非天然的位点鉴别序列(例如,标签或条型码);(c)将杂交复合物与延伸酶和dntp接触,其中对于每个杂交复合物,如果探查位置处的碱基与检测位置的碱基完美互补,则第一探针沿第二目标结构域延伸;(d)将延伸的第一探针与第二探针连接以形成扩增模板;(e)用第一和第二通用引物扩增所述扩增模板以产生扩增子,其中至少一个引物包括等位基因鉴别序列(例如,标签或条形码),其中等位基因鉴别序列包括无碱基位点;(f)将扩增子与切口核酸内切酶接触,从而在第二引物序列中产生3'突出;和(g)使用本文所述的目标多核苷酸表征方法,检测不同扩增子的位点鉴别序列和等位基因鉴别序列的存在,借此表明样品中所关心的不同靶标序列的存在。
[0406]
如本文所使用的,短语“多路”或语法等价形式是指不止一个所关心的目标序列的检测、分析或扩增。在一个实施方式中,多路是指至少100或200个不同的目标序列,同时至少500个不同的靶标序列是优选的。更优选地,至少1000个,特别优选地,5000或10,000个,并且最优选地,大于50,000或100,000个。检测可以在如本文所述的多个平台上进行。
[0407]
在一些方面,本文的发明公开提供了检测样品中核酸目标序列的方法。如本领域技术人员将理解的,样品溶液可以包含多种东西,其包括(但不限于)体液(包括(但不限于)实际上任何生物的血液、尿液、血清、淋巴、唾液、肛门和阴道分泌物、汗液和精液,并且哺乳动物样品是优选的,并且人样品是特别优选的);环境样品(包括(但不限于)空气、农业水和土壤样品);生物战剂样品;研究样品;纯化样品,如纯化的基因组dna、rna、蛋白质等;粗样品(细菌、病毒、基因组dna等)。如本领域技术人员将理解的,实际上可以对样品进行任何实验操作。
[0408]
如果需要,使用已知技术制备目标多核苷酸。例如,可以使用已知的裂解缓冲液、超声、电穿孔等处理样品使细胞裂解,并根据需要,如以下所列进行纯化和扩增,如本领域技术人员将理解的。另外,可以以多种方式进行本文所列的反应,如本领域技术人员将理解的。可以以任何顺序同时或顺序添加反应组分,其中下文列出了优选实施方式。另外,反应可以包括在测定中可以包含的多种其它试剂。这些包括试剂,如盐、缓冲剂、中性蛋白质,例
如白蛋白、去污剂等,其可以用于辅助最优化杂交和检测,和/或降低非特异性或背景相互作用。另外,根据样品制备方法和目标纯度,可以使用改善测定效率的试剂,如蛋白酶抑制剂、核酸酶抑制剂、抗微生物剂等。
[0409]
另外,在大部分实施方式中,将双链目标多核苷酸变性以使它们成为单链,从而允许引物和本文所述的其它探针杂交。一个实施方式使用了热步骤,通常通过将反应温度升高至约95℃,尽管还可以使用ph变化及其它技术。
[0410]
如本文所列,目标多核苷酸可以是反应产物,如来自反应的检测序列、连接的探针、来自pcr反应的延伸探针或pcr扩增产物(“扩增子”)等。
[0411]
在一些实施方式中,目标多核苷酸包括需要序列信息的位置,其在本文中一般地称为“检测位置”。在某一实施方式中,检测位置是单个核苷酸,尽管在一些实施方式中,它可以包括彼此邻接或被一个或多个核苷酸分开的多个核苷酸。如本文所使用的“多个”表示至少两个。如本文所使用的,杂交中与检测位置碱基碱基配对的碱基被称为“读取位置”或“审查位置”;因此,本发明的第一或第二步骤探针中的多个包含审查位置。
[0412]
本文所公开的方法可以采取多种构型,如附图中所示和如本文中更详细描述的。通常,这些部分包括复杂性减小部分(complexity reduction component)、特异性部分和扩增部分。可以以如下所公开的多种方式配置部分。也就是说,在一个实施方式中,首先执行复杂性减小步骤。随后是扩增或特异性步骤。作为另外一种选择,首先执行特异性步骤。随后可以是复杂性减小或扩增步骤。作为另外一种选择,首先执行扩增。随后是复杂性和特异性步骤。
[0413]
然而,以上表明可以以任何顺序执行三个部分中的每一个。本领域技术人员将理解当首先执行扩增时,将有可能涉及一定程度的复杂性减小或特异性。另外,当首先执行特异性部分时,将有一定程度的复杂性减小。另外,在一些实施方式中,当首先执行扩增时,将有一定程度的特异性和复杂性减小。然而,如下所述,所述方法通常包括三个部分。
[0414]
探针和引物
[0415]
如本领域技术人员所理解的,存在一些可以在本文所公开的方法中使用的探针或引物。这些探针/引物可以采取多种构型,并且可以具有以下更详细描述的多种结构成分。第一步骤探针可以是等位基因特异性探针或位点特异性探针。“等位基因特异性”探针或引物是指与目标序列杂交并且区分等位基因或与目标序列杂交并且以等位基因特异的方式修饰的探针或引物。“位点特异性”探针或引物是指以位点特异性方式杂交至目标序列,但不必需区分等位基因的探针或引物。如下所述,还可以修饰(即延伸)位点特异性引物,从而它包含了特定等位基因相关的信息,但是位点特异性引物不区分等位基因。
[0416]
在多种实施方式中,探针或引物包含一个或多个通用引发位点和/或鉴别序列。例如,在一种构型中,四个等位基因碱基中的每一个与不同序列(即等位基因鉴别序列(例如,标签或条型码))结合,每个序列具有类似的扩增效率。在另一个构型中,探针之一包含位点鉴别序列(例如,标签或条型码)。
[0417]
引物和探针核酸的大小可以不同,如本领域技术人员将理解的,探针的每个部分和探针的总长度一般在5至500个核苷酸长度改变。根据用途和扩增技术,每个部分可以在10至300、15至250或10至35个核苷酸长度之间。因此,例如,探针的通用引发位点可以在15
‑
20个核苷酸长度之间,在某些实施方式中,使用了18个核苷酸长度。探针的位点和/或等位
基因鉴别序列可以在10
‑
300个核苷酸长度之间,在某些实施方式中,使用了20
‑
100个核苷酸长度。探针的目标特异性部分可以在15
‑
50个核苷酸长度。另外,引物可以包含额外的扩增引发位点。
[0418]
在一个实施方式中,等位基因或位点特异性探针包含与目标序列的第一结构域基本互补的目标结构域。一般地,可以将探针设计成与目标序列(样品的目标序列或其它探针序列,如本文所述)互补,从而发生目标和本文所述的探针的杂交。这种互补性不必需是完全的;可以存在多个防碍本发明的目标序列和单链核酸之间杂交的碱基对错配。然而,如果突变数目过大从而即使在最不严格的杂交条件下也不能发生杂交时,所述序列与目标序列不互补。因此,本文中“基本互补”表示探针与目标序列足够互补,从而在所选反应条件下杂交。
[0419]
另外,可以构建在本文所述的方法中使用的探针以含有必需的引发位点或用于后续扩增方案的位点。在某些实施方式中,所述引发位点是通用引发位点。本文中“通用引发位点”或“通用引发序列”是指将结合用于扩增的引物的探针序列。
[0420]
如本领域技术人员将理解的,一般地,可以实施高度多路反应,对于所有反应,所有通用引发位点相同。作为另外一种选择,可以同时或顺序使用通用引发位点“组”和相应探针。通用引发位点用于扩增修饰的探针以形成多个扩增子,然后以多种方式检测扩增子,如本文所列。
[0421]
因此,本文所述的方法提供了第一目标探针组。本文中“探针组”是指在特定多路测定中使用的多个目标探针。在本文中,多个表示至少2个,其中根据测试的测定、样品和目的,优选大于10个。在一个实施方式中,探针组包括大于100个探针,优选大于500个探针,并且更优选大于1000个探针。在特别优选的实施方式中,每个探针组含有至少5000个探针,最优选大于10,000个探针。
[0422]
复杂性减小部分
[0423]
复杂性减小可以是本文所述的多路方案的部分。通常,复杂性减小是富集特定目标或位点的方法。也就是说,认为复杂性减小是导致非目标核酸从样品中移除或未正确杂交或根本未杂交至目标核酸的探针/引物的移除的方法。另外,复杂性减小包括酶促步骤期间未修饰的探针的移除。也就是说,复杂性减小包括在酶促步骤(即扩增或特异性步骤或两者)之前移除非目标核酸,即富集目标核酸,或移除未杂交的探针或引物。
[0424]
存在多个包含复杂性减小步骤的方法。这些包括(但不限于)以目标特异性方式修饰的目标核酸或探针/引物的选择性固定化,非目标核酸的选择性移除和非目标核酸的选择性破坏。这种破坏包括(但不限于)非目标核酸的变性、降解或切割。另外,复杂性减小可以包括如目标选择性扩增的部分,尽管这还包括扩增和部分。
[0425]
在某些实施方式中,复杂性减小伴随着以目标特异性方式选择性固定修饰的引物。也就是说,位点特异性或等位基因特异性引物与目标杂交。目标可以固定化或处于溶液中。杂交后,引物在引物延伸反应中延伸。在一些方面,引物或ntp包含纯化标签,其使得能够从反应混合物中移除或纯化延伸产物。一旦延伸,通常可以将修饰的引物固定在固体载体上。修饰的引物固定化后,可以清洗载体以除去未修饰(即延伸)的非目标核酸和引物两者。因此,固定化的引物包括目标位点相关信息,其包括特定等位基因信息。这导致目标核酸的富集或非目标核酸的移除。
[0426]
在另一个实施方式中,复杂性减小部分包括目标多核苷酸的选择性固定化。也就是说,优先地将目标多核苷酸而不是非目标核酸固定在固体载体上。
[0427]
在一个实施方式中,将目标多核苷酸、探针或引物,包括修饰的引物连接至固体载体。本文中“固体载体”或其它语法等价形式是指适合于或者可以修饰以适合于与目标序列连接的任何材料。如本领域技术人员将理解的,可能的底物数目非常大。可能的底物包括(但不限于)玻璃和修饰或官能化的玻璃、塑料(包括丙烯酸类、聚苯乙烯以及苯乙烯共聚物及其它材料、聚丙烯、聚乙烯、聚丁烯、聚氨脂、teflon
tm
等)、多糖、尼龙或硝化纤维素、陶瓷、树脂、二氧化硅或二氧化硅基材料,包括硅和修饰的硅、碳、金属、无机玻璃、塑料、光纤束和多种其它聚合物。磁珠和高通量微滴板是特别优选的。
[0428]
固体载体的组成和几何形状根据其用途而改变。在某些实施方式中,包括微球或珠的载体可以用于固体载体。本文中“微球”或“珠”或语法等价形式表示小的分离颗粒。根据生物活性剂的种类和合成方法,珠的成分将改变。适合的珠成分包括在肽、核酸和有机部分合成中使用的那些,其包括(但不限于)塑料、陶瓷、玻璃、聚苯乙烯、甲基苯乙烯、丙烯酸聚合物、顺磁性物质、二氧化钍溶胶、石墨化碳、二氧化钛、乳胶或交联右旋糖酐,如琼脂糖、纤维素、尼龙、交联胶束和聚四氟乙烯以及本文所列的用于固体载体的任何其它材料,所有材料均可以使用。来自bangs laboratories,fishers in的“microsphere detection guide”是有帮助的指南。优选地,在该实施方式中,当执行复杂性减小时,所述微球是磁性微球或珠。
[0429]
一旦连接到固体载体,对目标序列、探针或引物进行如本文所述的分析。
[0430]
在本发明中可以使用多种杂交或清洗条件,其包括高、中和低严格性条件;参见,例如maniatis等人,molecular cloning:a laboratory manual,第2版,1989和short protocols in molecular biology,ausubel等人主编,以上文献作为参考并入本文。严格性条件是序列依赖性的并且在不同情况下将是不同的。较长的序列在较高的温度下特异性杂交。核酸杂交的广泛指南可见于tijssen,techniques in biochemistry and molecular biology
‑
hybridization with nucleic acid probes,“overview of principles of hybridization and the strategy of nucleic acid assays”(1993)。通常,严格性条件选择为比限定离子强度和ph下特定序列的热熔点(tm)低约5
‑
10℃。tm是(限定离子强度、ph和核酸浓度下)50%与目标互补的探针杂交至目标序列达到平衡时的温度(由于在tm,目标序列过量存在,因此平衡时50%的探针被占据)。严格性条件将是其中ph 7.0至8.3,盐浓度小于约1.0m钠离子,通常约0.01至1.0m钠离子浓度(或其它盐),并且对于短探针(例如,10至50个核苷酸)温度至少约30℃,对于长探针(例如,大于50个核苷酸),至少约60℃的那些条件。还可以通过添加螺旋去稳定剂,如甲酰胺来实现严格性条件。
[0431]
本文中“延伸酶”是指通过加入ntp使序列延伸的酶。如本领域中熟知的,存在多种适合的延伸酶,其中聚合酶(rna和dna两者,基于目标序列组成和预环化探针(precircle probe))是优选。优选的聚合酶是缺乏链置换活性,从而它们将能够仅在探针末端添加必要的碱基,而不会进一步延伸探针以包含与目标结构域互补的核苷酸并因此防止环化的那些。适合的聚合酶包括(但不限于)dna和rna聚合酶两者,其包括dna聚合酶i的克列诺片段、sequenase 1.0和sequenase 2.0(u.s.biochemical)、t5 dna聚合酶、phi29 dna聚合酶和多种rna聚合酶,如来自栖热菌属(thermus sp.)的rna聚合酶,或来自噬菌体的qβ复制酶,
以及sp6、t3、t4和t7 rna聚合酶等均可使用。
[0432]
聚合酶还可以包括本质上缺少5'至3'核酸外切酶活性以确保探针将不会延伸通过探针的5'端的那些。缺少5'至3'核酸外切酶活性的示例性酶包括dna聚合酶的克列诺片段和dnaptaq聚合酶的stoffel片段。例如,taq dna聚合酶的stoffel片段由于基因操作而缺少5'至3'核酸外切酶活性,这导致缺少n末端289个氨基酸的截短蛋白的产生。(参见,例如,lawyer等人,j.biol.chem.,264:6427
‑
6437(1989);和lawyer等人,pcr meth.appl.,2:275
‑
287(1993))。对于来源于海栖热袍菌(t.maritima)的聚合酶、tsps17、tz05、tth和taf,已产生了类似的突变体聚合酶。
[0433]
其它聚合酶是缺少3'至5'核酸外切酶活性的那些,这种活性通常被称为校对活性并且它移除了引物模板双螺旋3'端的错配碱基。尽管3'至5'核酸外切酶活性的存在提供了链合成中提高的保真度,但是热稳定的dna聚合酶(如tma,包括缺少5'至3'核酸外切酶活性的tma突变形式)中存在的3'至5'核酸外切酶活性也降解单链dna,如pcr中使用的引物、单链模板和单链pcr产物。引物延伸过程中使用的寡核苷酸引物的3'端的完整性至关重要,这只因为正是从该末端开始新生链的延伸。3'端的降解导致寡核苷酸缩短,这反过来导致引发反应中特异性的丧失(即引物越短,则越有可能发生伪引发或非特异性引发)。
[0434]
其它聚合酶是热稳定性聚合酶。耐热酶可以包括在最优条件下,在40℃保持1小时后仍保留其大部分活性的任何酶。缺少5'至3'核酸外切酶和3'至5'核酸外切酶的热稳定性聚合酶的实例包括taq dna聚合酶的stoffel片段。该聚合酶由于基因操作而缺少5'至3'核酸外切酶活性,并且由于taq聚合酶天然缺少3'至5'核酸外切酶活性,因此不存在3'至5'活性。tth dna聚合酶衍生自嗜热栖热菌(thermus thermophilus),并且可得自epicentre technologies,molecular biology resource inc.或perkin
‑
elmer corp.。其它有用的缺少3'核酸外切酶活性的dna聚合酶包括vent[r](exo
‑
),其得自new england biolabs,inc.(从带有来自古细菌thermococcus litoralis的dna聚合酶基因的大肠杆菌(e.coli)菌株纯化得到)和来源于黄栖热菌(thermus flavus)并且得自amersham corporation的hot tub dna聚合酶。其它优选的热稳定并且缺少5'至3'核酸外切酶活性和3'至5'核酸外切酶活性的酶包括amplitaq gold。可以使用至少基本相同的其它dna聚合酶,如其它n
‑
末端截短的水生栖热菌(thermus aquaticus)(taq)dna聚合酶i。称为klentaq i和klentaq la的聚合酶非常适合该目的。当然,根据本发明还可以使用具有这些特性的任何其它聚合酶。
[0435]
在探针3'端添加一个或多个核苷酸的条件将取决于所使用的特定的酶,并且通常将按照所使用的酶的生产商所推荐的条件进行。
[0436]
特异性部分
[0437]
一般在复杂性减小步骤之后,在本文所述的方法中包含特异性步骤。“特异性部分”是指区分目标核酸的步骤,优选地,在等位基因电平。也就是说,特异性部分是等位基因特异性步骤(例如,基因分型或snp分析)。尽管可以通过将等位基因特异性探针与模板(即以上复杂性减小步骤的产物)简单杂交来达到一定特异性电平,但是在优选的实施方式中,特异性步骤包括酶促步骤。也就是说,酶促步骤的保真度改善了等位基因区分的特异性。优选的酶包括如本文中更详细地描述的dna聚合酶、rna聚合酶和连接酶。
[0438]
如上所述的聚合酶还可以适合于特异性步骤。
[0439]
多种连接酶是已知的并且适合于在本文所述的方法中使用。在lehman,science,
oligonucleotide)。通过探查到的不同的snp等位基因的数目确定寡核苷酸的数目。例如,如果探查到1000个snp,则每个具有两个等位基因,因此2000个寡核苷酸将是必需的。询问子(interrogator)与含有snp的dna延伸互补,其中每个询问子的末端碱基对应于snp位置,或者其中snp
‑
特异性位置位于询问子的最后1、2、3或4个核苷酸内。在一些实施方式中,询问子不是引物的末端位置,而是位于距引物3'端1、2、3、4、5或6个核苷酸的位置处。例如,当snp具有a和c等位基因时,提供了以t和g结尾的询问子,并且在一些实施方式中,可以将其固定在单独的元件(珠)上以检测所述两个等位基因。尽管匹配和错配均将杂交至给定等位基因,但是仅匹配可以用作dna聚合酶延伸反应的引物。因此,在探针与目标dna杂交后,进行聚合酶反应。这导致在存在dntp的情况下,与dna聚合酶的杂交物的延伸。
[0446]
在某些实施方式中,由于未延伸的探针或引物可以与延伸引物在捕获探针结合中竞争,因此期望从测定混合物,并且具体地,从固体载体中除去未延伸或未反应的探针或引物。相对于延伸引物,未延伸的引物的浓度可以相对较高,这是因为通常需要大量过量的引物以产生有效的引物退火。因此,可以使用一些不同的技术来辅助未延伸的探针或引物的除去。这些通常包括基于通过结合至固体载体,保护反应的引物并降解未延伸的引物,和分离未反应和反应的引物除去未反应的引物的方法。
[0447]
扩增部分
[0448]
在该实施方式中,本文提供了可以在表征多核苷酸的方法中使用的包括多核苷酸扩增的方法以及核酸扩增反应的产物(即扩增子)。适合的扩增方法包括目标扩增和信号扩增。目标扩增包括要检测的目标序列的扩增(即复制),从而导致目标分子数目的显著增加。目标扩增策略包括(但不限于)聚合酶链反应(pcr)、链置换扩增(sda)、核酸序列基扩增(nasba)和滚环扩增(rca)。这些扩增策略对本领域技术人员来说是熟知的,并且可以容易地选择用于所述方法。
[0449]
作为另外一种选择,替代技术使用目标作为模板来复制信号探针,而不是扩增目标,这使得少量目标分子能够导致产生大量信号检验,然后可以进行检测。信号扩增策略包括连接酶链反应(lcr)、循环探针技术(cpt)、侵入性裂解技术,如invader技术、q
‑
β复制酶(qr)技术和“扩增探针”,如“分支dna”的使用,其导致产生了结合单个目标序列的多重标记探针。
[0450]
所有这些方法都可以包含引物核酸(包括核酸类似物),其杂交至目标序列以形成杂交复合物,并且以修饰引物形成修饰的引物的方式添加酶。例如,pcr通常需要两种引物,dntp和dna聚合酶;lcr需要两种引物,其邻近杂交目标序列和连接酶;cpt需要一种可切割引物和切割酶;侵入性裂解需要两种引物和裂解酶;等。因此,一般地,将目标核酸加入至包含必要扩增部分的反应混合物,并形成修饰的引物。
[0451]
一般地,将修饰的引物用作二次反应的目标序列,然后其产生多个扩增链,并且可以如本文所列进行检测。根据需要,以多种方式除去未反应的引物,如本领域技术人员将理解的和如本文所列的。因此,反应从向目标序列中加入引物核酸开始,其形成了杂交复合物。一旦引物和目标序列之间形成杂交复合物,则使用酶(有时称为“扩增酶”)修饰引物。如对于本文所列的所有方法,可以在测定期间的任意点(在加入引物之前、期间或之后)加入酶。酶的身份将取决于所使用的扩增技术。类似地,修饰将取决于扩增技术。
[0452]
在某些实施方式中,目标扩增技术是聚合酶链反应(pcr)。pcr是广泛使用和描述
的,并且包括引物延伸与热循环结合使用以扩增目标序列;参见美国专利no.4,683,195和4,683,202和pcr essential data,j.w.wiley&sons,ed.c.r.newton,1995,以上全部文献作为参考并入本文。另外,存在多个pcr的变化,其也在本发明中得到应用,包括“定量竞争性pcr”或“qc
‑
pcr”、“任意引物pcr”或“ap
‑
pcr”、“免疫
‑
pcr”、“alu
‑
pcr”、“pcr单链构象多态性”或“pcr
‑
sscp”、“反转录酶pcr”或“rt
‑
pcr”、“生物素俘获pcr”、“小载体pcr(vectorette pcr)”、“锅饼式(panhandle)pcr”和“pcr选择cdna减少(subtraction)”、“等位基因
‑
特异性pcr”等。应理解本领域技术人员可以容易地选择可以在本文所述的方法中使用的pcr的适当改变。
[0453]
在某些实施方式中,扩增反应是如本文所述的多路扩增反应。在一个实施方式中,扩增反应使用多个pcr引物来扩增多个靶标序列。在该实施方式中,使用多个扩增引物对同时扩增多个目标序列。
[0454]
在替代实施方式中,多路pcr反应使用如本文所述的通用引物。也就是说,通用pcr引物杂交至目标序列上的通用引发位点并借此扩增多个靶标序列。该实施方式潜在地是优选的,这是因为它仅需要有限的pcr引物。也就是说,少至一个引物对可以扩增多个靶标序列。
[0455]
如前所述,使用人dna作为模板产生了golden gate扩增子(cold spring harb symp quant biol.2003;68:69
‑
78.highly parallel snp genotyping.fan jb等人)。基于等位基因,所得扩增子具有表示为p1和p2的两个引物之一。此外,在所有扩增子上存在通用反向引物(“反向p3”)。
[0456]
p1:tctcgtcgctcatcaact(seq id no:82)
[0457]
p2:gagtcgaggtcatatcgt(seq id no:83)
[0458]
反向p3:gtctgcctatagtgagtc(seq id no:84)
[0459]
将使用16个循环的第二轮pcr用于添加称为“p1_barcode_a”和“p1_barcode_b”的等位基因条型码引物。使用含有多个脱氧尿嘧啶残基的延伸的通用反向引物(“universal du reverse”)。
[0460]
p1_barcode_a:
[0461]
/5phos/tttttttttttttttccttccxxttttcttcttcttcaagaagaagatctcgtcgctcatcaact(seq id no:85)
[0462]
p2_barcode_b:
[0463]
/5phos/tttttttttttttttccttccxxttttttaattaattttgttgttgtgagtcgaggtcatatcgt(seq id no:86)
[0464]
universal du reverse:
[0465]
atacggcg/du/ccaccgacc/du/cagcgtc/du/gcctatag/du/gagtc(seq id no:87)
[0466]
其中/5phos/表示5'磷酸盐,/du/是脱氧尿嘧啶残基,并且x是无碱基部分。
[0467]
pcr之后,将样品与user酶(new england biolabs,ipswich,ma)在37℃培育2.5小时以产生单链缺口,在此存在du残基。将样品加热至65℃,10min以除去碎片化的dna并产生3'突出。使用pcr cleanup试剂盒(qiagen)纯化样品。
[0468]
通过加热至65℃并缓慢冷却,将样品与含有胆固醇的低聚“p3_chol”以1:1的摩尔比退火。
[0469]
p3_chol:
[0470]
accgacactgcgtctgcctatagtgagtc/isp9//3cholteg/(seq id no:88)
[0471]
其中/isp9/表示9
‑
原子三甘醇间隔臂,并且/3cholteg/表示3'胆固醇teg(三甘醇)部分。
[0472]
脂质双分子层由1,2
‑
二植烷酰基
‑
sn
‑
甘油基
‑3‑
胆碱磷酸(avanti极性脂)形成。双分子层在teflon中横跨电平直径~20微米的孔。以~2.5ng/ml的浓度,将m2
‑
nnn
‑
mspa加入至双分子层的底侧(grounded side)。一旦插入单个孔,则隔室内冲入实验缓冲液以避免进一步插入。axopatch
‑
200b膜片钳放大器(axon instruments)将180mv的电压施加到双分子层两侧并测量离子电流。使用4
‑
极贝塞耳滤波器以50khz低通滤波模拟信号,然后以低通滤波频率的5倍数字化。使用以labwindows/cvi写入的常规软件(national instruments)控制数据采集。
[0473]
双分子层两侧~60μl的隔室含有0.4m kcl、1mm edta、1mm dtt、1mm atp、10mm mgcl2和10mm hepes/koh缓冲液,ph 8.0的实验缓冲液。以150nm,将hel308野生型tga用作马达。
[0474]
图23示出了根据一些实施方式对于适合用作各自条型码的第一说明性多核苷酸序列(seq id no:89)和第二说明性多核苷酸序列(seq id no:90),可以作为时间函数产生的示例性模拟信号。在图24中可以看出(1)对应于第一说明性多核苷酸序列通过hel308解旋酶通过孔的分步易位的模拟信号具有随时间的特征图形,其包括相对高信号电平的两个“峰”,然后下降,然后是相对低信号电平的另外两个“峰”,而(2)对应于第二说明性多核苷酸序列通过hel308解旋酶通过孔的分步易位的模拟信号包括相对低信号电平的两个“峰”,然后升高,然后是相对高信号电平的另外两个“峰”。因此,可以预期可以(例如)使用图形匹配来容易地彼此区分包括不同特征的真实信号,如模拟信号(1)和(2),并因此可以有利于区分彼此的测定结果。例如,图24a
‑
24d示出了根据一些实施方式对于适合用作各自条型码的第一和第二说明性多核苷酸序列可以作为时间函数产生的示例性模拟信号。可以看出在图24a和24b中通常表示为“条形码”的模拟信号的部分包括相对高信号电平的两个“峰”,然后下降,然后是相对低信号电平的另外两个“峰”,并因此可以理解为对应于第一说明性多核苷酸序列。还可以看出在图24c和24d中通常表示为“条形码”的模拟信号的部分包括相对低信号电平的两个“峰”,然后升高,然后是相对高信号电平的另外两个“峰”,并因此可以理解为对应于第二说明性多核苷酸序列。
[0475]
在另一个实例中,以与本实施例中如上所述的类似方式,将2nnn mspa孔插入dphpc脂质双分子层。缓冲液包括400mm kcl、10mm hepes ph 8、5mm mgcl2和1mm edta。所述试剂包括1mm dtt和1mm atp。酶包括约150mm的hel308 tga。dna为约10nm,并且将测序的单链(表示rs1801131 snp1和snp2)杂交至含有胆固醇的多核苷酸。使用后处理解释这些链测序期间所获得的信号,其包括电平查找(level
‑
finding)和使用算法与预期序列比对,如在本文其它处所述的。
[0476]
图25a和25b分别示出了根据一些实施方式对于适合用作各自条型码的第一和第二说明性多核苷酸序列可以作为时间函数产生的示例性模拟信号。图25a和25b中的虚线框内的模拟信号部分对于分别表示为rs1801131snp1和rs1801131snp2的序列分别包括了不同的图像,并因此可以用作各自的条型码。用作条型码的序列与图23中所示的相同。
[0477]
图26a
‑
26d分别示出了根据一些实施方式对于适合用作各自条型码的第一和第二说明性多核苷酸序列作为时间函数产生的示例性测量信号。图26a和26b中的虚线框内的测量信号部分分别包括不同的图形,可以看出其对应于表示为rs1801131snp1的序列条型码,而图26c和26d中的虚线框内的测量信号部分分别包括不同的图形,可以看出其对应于表示为rs1801131snp2的序列条型码,并且该条形码与表示为rs1801131snp1的序列的条型码是容易地可区分的。
[0478]
作为参考并入
[0479]
在整个发明申请中,使用或未使用括号引用了多个出版物。在本发明申请中,这些出版物的公开内容出于所有目的以它们的全部内容作为参考并入本文,其包括(但不限于)为了更充分地描述本发明公开所述领域目前的工艺水平。
[0480]
其它替代实施方式
[0481]
应注意可以使用执行指令(例如,软件命令)以实施本文所公开的操作的多种类型的数据处理器环境(例如,在一个或多个数据处理器上)来实施本文所提供的系统和方法。非限制性实例包括在单个通用计算机或工作站上,或者在网络系统上,或者在客户
‑
服务器配置中,或者在应用业务供应商配制中实施。例如,可以在多种不同类型的处理装置上,通过包含设备处理子系统可执行的程序命令的程序代码来实施本文所述的方法和系统。软件程序命令可以包括源编码、结果代码、机器代码或者可操作以导致处理系统执行本文所述的方法和操作的任何其它存储数据。然而,还可以使用其它装置,如配置用于执行本文所述的方法和系统的固件或甚至适当设计的硬件。
[0482]
还应注意所述系统和方法可以包括通过用于一个或多个数据处理装置通讯的网络(例如,局域网、广域网、因特网、它们的组合等)、光学纤维介质、载波、无线网络等传输的数据信号。数据信号可以具有任何或所有本文所公开的提供给装置或装置所提供的数据。
[0483]
可以在一种或多种不同类型的计算机实施数据存储中存储和实施系统和方法数据(例如,关联、数据输入、数据输出、中间数据结果、最终数据结果等),如不同类型的存储设备和编程构造体(例如,ram、rom、闪存、平面文件、数据库、编程数据结构、编程变量、if
‑
then(或类似类型)声明构造体等)。注意数据结构描述了在供计算机程序使用的数据库、程序、存储器或其它计算机可读介质中组织和存储数据中使用的格式。
[0484]
还可以在多种不同类型的计算机可读存储介质上提供系统和方法,其包括含有在通过处理器的执行中用于实施方法操作和执行本文所述的系统的命令(例如,软件)的计算机存储机制(例如,永久性介质,如cd
‑
rom、磁盘、ram、闪存、计算机硬盘等)。
[0485]
此外,本文所提供的计算机组件、软件模块、功能、数据存储和数据结构可以直接或间接彼此连接以使得能够进行它们的操作所需的数据流动。还注意模块或处理器包括(但不限于)执行软件操作的代码单元,并且可以(例如)作为代码的子例程单元或作为代码的软件功能单元或作为对象(如在面向对象的范例中那样)或作为小程序或以计算机脚本语言或作为另一类型的计算机代码实施。基于实际情形,可以将软件部件和/或功能定位在单个计算机上或分布在多个计算机上。
[0486]
尽管已参考所公开的实施方式描述了本发明公开,但是本领域技术人员将容易地理解以上详述的具体实例和研究仅是本发明公开的说明。应理解在不背离本发明公开的精神的情况下,可以做出多种改变。因此,本发明公开仅限于所附权利要求。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1