一种尼拉帕尼的制备方法与流程
1.本发明属于生物医药技术领域,尤其是涉及一种尼拉帕尼的制备方法。
背景技术:
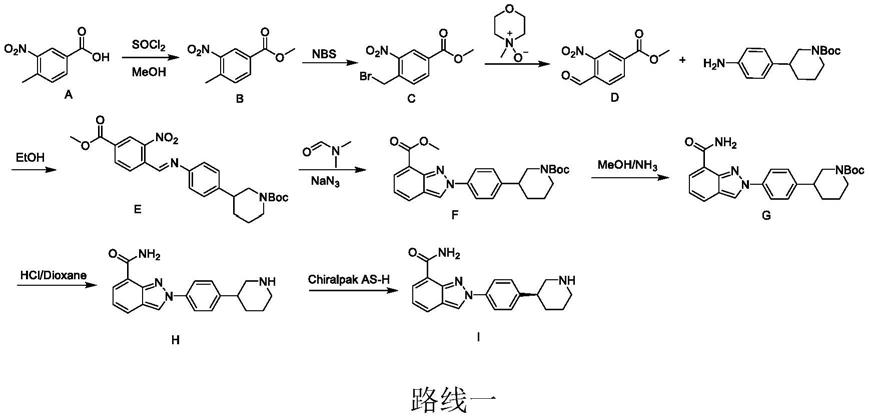
2.尼拉帕尼是一种治疗卵巢恶性肿瘤的靶向药,是多聚腺苷二磷酸核糖聚合酶抑制剂,是继奥拉帕尼以后,fda批准的第三个parp抑制剂,为卵巢癌的治疗增加了新的选择。尼拉帕尼的作用是修复dna双链损伤,保证细胞内dna损伤后得到及时修复以避免发生癌变。现有的尼拉帕尼合成技术中的专利文献包括discovery of 2
‑
{4
‑
[(3s)
‑
piperidin
‑3‑
yl]phenyl}
‑
2h
‑
indazole
‑7‑
carboxamide(mk
‑
4827):a novel oral poly(adp
‑
ribose)polymerase(parp)inhibitor efficacious in brca
‑
1and
‑
2mutant tumors,development of a fit
‑
for
‑
purpose large
‑
scale synthesis of an oral parp inhibitor等,公开了一些尼拉帕尼的合成路线1‑2。
[0003][0004]
该合成路线以3
‑
甲基
‑2‑
硝基苯甲酸为起始原料,经8步反应得到目标化合物,其反应路线长,且用到叠氮化钠等危险试剂。对环境产生巨大污染,工艺当中存在危险因素,且产率不高。
[0005][0006]
路线二合成方法首先通过手性拆分的方式得到(s)
‑3‑
(4
‑
氨基苯基)哌啶后与3
‑
甲酰基
‑
2硝基苯甲酸甲酯缩合,并在叠氮化钠的作用下构建了吡唑环。该路线在还原吡啶环时用了二氧化,铂成本较高,且也用到叠氮化钠危险试剂,不利于放大。
技术实现要素:
[0007]
为解决上述问题,本发明提出了一种合成工艺路线简单、制备效率高、对人体和环境损害小、有效降低合成成本的尼拉帕尼的制备方法。
[0008]
为达到上述目的,本发明的技术方案是这样实现的:
[0009]
一种尼拉帕尼的制备方法,包括如下合成路线:
[0010][0011]
其中,步骤a为:化合物1和溴苯在pd催化剂光催化下,得到化合物2,即得到尼拉帕尼关键中间体;
[0012]
步骤b和步骤c为:化合物2经过手性拆分,得到化合物4;
[0013]
步骤d为:化合物4和化合物5在溴化铜催化下进行偶联,得到化合物6,即得到保护的尼拉帕尼;
[0014]
步骤e为:化合物6在甲磺酸作用下脱去保护色,在四氢呋喃打浆下得到化合物7,即得到目标产物尼拉帕尼。
[0015]
进一步的,所述步骤a包括:将化合物1和pd催化剂溶于溴苯中;加入甲酸钾,混匀后得到橙色浑浊溶液;对橙色浑浊溶液进行照射,配合搅拌,至hplc显示化合物1反应完全;
反应结束后,进行后处理得到化合物2;
[0016]
所述化合物2为1
‑
[3
‑
(4
‑
溴苯基)哌啶
‑1‑
基]
‑
2,2二甲基丙烷
‑1‑
酮,为npv哌啶类衍生物,其的化学式为:
[0017][0018]
进一步的,所述步骤b包括:化合物2在盐酸条件下脱去保护;用d
‑
(+)
‑
二苯甲酰酒石酸、d
‑
酒石酸或d
‑
樟脑磺酸中的任意一种进行手性拆分,得到化合物3。
[0019]
进一步的,所述步骤c包括:化合物3通过pvcl保护仲胺,得到化合物4。
[0020]
进一步的,所述步骤d包括:化合物4和化合物5在溴化铜催化下,用8
‑
羟基喹啉做配体,在105℃~115℃条件下进行偶联,得到化合物6;所述化合物5为nboc
‑
1h
‑
吲唑
‑7‑
羧酰胺。
[0021]
进一步的,所述pd催化剂为pd(pph3)4,pd(oac)2,pd2(dba)3,pd(dppf)cl2,pd(pph3)2cl2或pdcl2中的任意一种。
[0022]
上述催化剂在受到光照时,辐射的光子会激发pd催化剂,发生单电子转移,从而产生了烷基自由基,对底物进行自由基加成反应,经后续反应得到尼拉帕尼关键中间体。
[0023]
进一步的,所述步骤a中,照射所用的光源为可见光或波长为450nm
‑
480nm的光或波长为500nm
‑
560nm的光。
[0024]
进一步的,所述步骤a中,对橙色浑浊溶液进行照射时的反应温度为20℃~60℃。
[0025]
进一步的,所述步骤a中的进行后处理具体为:采用浓缩处理除去溶剂,正己烷和乙酸乙酯重结晶,得到尼拉帕尼关键中间体;或采用饱和食盐水进行洗涤,并用乙酸乙酯进行萃取,萃取完成后合并有机相,采用无水硫酸钠进行干燥浓缩,利用硅胶柱柱层析,得到尼拉帕尼关键中间体。
[0026]
相对于现有技术,本发明所述的尼拉帕尼的制备方法具有以下优势:
[0027]
(1)本发明所述的尼拉帕尼的制备方法合成工艺简单高效,合成成本低,无高温高压反应,适用的温度较广,反应条件简单易得;
[0028]
(2)本发明所述的尼拉帕尼的制备方法采用pd催化剂,利用洁净的光能源产生自由基反应,反应活性好,选择性高,可以替代危险试剂的使用,对环境友好,不会损害人身体健康,可以高效的实现目标化合物的构建,并且产生的废物较少,原子利用率较高,符合绿色化学的概念,同时可以有效降低尼拉帕尼的合成成本,在环保要求越来越严格的工业化生产中,将占领技术领先地位。
具体实施方式
[0029]
除有定义外,以下实施例中所用的技术术语具有与本发明所属领域技术人员普遍理解的相同含义。以下实施例中所用的试验试剂,如无特殊说明,均为常规生化试剂;所述实验方法,如无特殊说明,均为常规方法。
[0030]
下面结合实施例来详细说明本发明。
[0031]
一种尼拉帕尼的制备方法,包括如下步骤:
[0032]
s1、将化合物1和pd催化剂(0.05eq)溶于溴苯中,加入甲酸钾(1.2eq),混匀后得到橙色浑浊溶液;
[0033]
其中,化合物1的化学式为pd催化剂为pd(pph3)4,pd(oac)2,pd2(dba)3,pd(dppf)cl2,pd(pph3)2cl2或pdcl2中的任意一种;
[0034]
s2、对橙色浑浊溶液进行照射,配合搅拌,至hplc显示化合物1反应完全;
[0035]
其中,照射所用的光源为可见光或波长为450nm
‑
480nm的光或波长为500nm
‑
560nm的光;反应温度为20℃~60℃;
[0036]
s3、反应结束后,进行后处理得到化合物2,即尼拉帕尼关键中间体;
[0037]
其中,后处理具体为:采用浓缩处理除去溶剂,正己烷和乙酸乙酯重结晶,得到尼拉帕尼关键中间体;或采用饱和食盐水进行洗涤,并用乙酸乙酯进行萃取,萃取完成后合并有机相,采用无水硫酸钠进行干燥浓缩,利用硅胶柱柱层析,得到尼拉帕尼关键中间体。
[0038]
所述的尼拉帕尼关键中间体为1
‑
[3
‑
(4
‑
溴苯基)哌啶
‑1‑
基]
‑
2,2二甲基丙烷
‑1‑
酮,其化学式为:
[0039][0040]
s4、化合物2在盐酸条件下脱去保护;用d
‑
(+)
‑
二苯甲酰酒石酸、d
‑
酒石酸或d
‑
樟脑磺酸中的任意一种进行手性拆分,得到化合物3;
[0041]
化合物3的化学式为:
[0042]
s5、化合物3通过pvcl保护仲胺,得到化合物4;
[0043]
化合物4的化学式为:
[0044]
s6、化合物4和化合物5在溴化铜催化下,用8
‑
羟基喹啉做配体,在105℃~115℃条件下进行偶联,得到化合物6;
[0045]
所述化合物5为nboc
‑
1h
‑
吲唑
‑7‑
羧酰胺,其化学式为:
[0046][0047]
所述化合物6为保护的尼拉帕尼,其化学式为:
[0048][0049]
s7、化合物6在甲磺酸作用下脱去保护色,在四氢呋喃打浆下得到化合物7,即得到目标产物尼拉帕尼;
[0050]
所述化合物7的化学式为:
[0051]
具体合成路线如下所示(催化剂以pd(pph3)4为例):
[0052][0053]
1.制备化合物2,即尼拉帕尼关键中间体1
‑
[3
‑
(4
‑
溴苯基)哌啶
‑1‑
基]
‑
2,2二甲基丙烷
‑1‑
酮
[0054]
实施例1
[0055]
将化合物1(10.0g,40.0mmol)与pd(pph3)4(0.94g,0.80mmol)溶解于溴苯(300ml)中,加入甲酸钾(4.20g,50.0mmol),得到一橙色浑浊溶液;随后,在可见光照射下,加热至50℃,搅拌24h,hplc显示化合物1反应完全;反应结束后,用饱和的氯化钠(300ml)洗涤,用乙酸乙酯(3
╳
200ml)萃取,合并有机相,用无水硫酸钠干燥,浓缩,利用硅胶柱柱层析(石油醚/乙酸乙酯),得到尼拉帕尼关键中间体为白色固体(6.97g,54%),熔点测试62.5℃~63.1℃。
[0056]
实施例2
[0057]
将化合物1(10.0g,40.0mmol)与pd(pph3)2cl2(0.56g,0.80mmol)溶解于溴苯
(300ml)中,加入甲酸钾(4.20g,50.0mmol),得到一橙色浑浊溶液;随后,在可见光照射下,加热至50℃,搅拌24h,hplc显示第一中间体反应完全;反应结束后,用饱和的氯化钠(300ml)洗涤,用乙酸乙酯(3
╳
200ml)萃取,合并有机相,用无水硫酸钠干燥,浓缩,利用硅胶柱柱层析(石油醚/乙酸乙酯),得到尼拉帕尼关键中间体为白色固体(5.67g,44%),熔点测试62.5℃~63.1℃。
[0058]
实施例3
[0059]
将化合物1(10.0g,40.0mmol)与pd(oac)2(0.18g,0.80mmol)溶解于溴苯(300ml)中,加入甲酸钾(4.20g,50.0mmol),得到一橙色浑浊溶液;随后,用可见光照射下,加热至50℃,搅拌24h,hplc显示第一中间体反应完全;反应结束后,用饱和的氯化钠(300ml)洗涤,用乙酸乙酯(3
╳
200ml)萃取,合并有机相,用无水硫酸钠干燥,浓缩,利用硅胶柱柱层析(石油醚/乙酸乙酯),得到尼拉帕尼合成关键中间体为白色固体(6.58g,51%),熔点测试62.5℃~633.1℃。
[0060]
实施例1~3得到的尼拉帕尼关键中间体的化学式为:
[0061][0062]
对尼拉帕尼关键中间体进行检测得到以下数据:
[0063]
h nmr(400mhz,chloroform
‑
d)δ7.44
–
7.25(m,2h),7.03
–
6.95(m,2h),4.39
–
4.07(m,2h),2.89
–
2.68(m,3h),1.97(d,j=9.1hz,1h),1.74(d,j=8.2hz,1h),1.49(d,j=10.5hz,2h),1.20(s,9h),
[0064]
13
c nmr(100mhz,chloroform
‑
d)δ153.9,142.1,130.3,127.0,121.4,40.9,38.9,31.0,29.0,26.4。
[0065]
2.制备尼拉帕尼
[0066]
实施例4
[0067]
制备化合物3:
[0068]
将实施例1~3制备的尼拉帕尼关键中间体(10.0g,30.0mmol)溶于120ml4n的盐酸二氧六环溶液中,30℃~40℃反应1h;随后反应液中加300ml饱和碳酸氢钠溶液,用200ml乙酸乙酯萃取,无水硫酸钠干燥,取滤液旋干,得到白色固体;室温下将粗品溶于150ml乙醇中,将d
‑
(+)
‑
二苯甲酰酒石酸(17.2g,48.0mmol)溶于150ml乙醇中缓慢滴加进体系中,加热至80℃,回流反应两小时;后降温至35~45℃左右,有固体析出,趁热抽滤,用少量乙酸乙酯洗涤滤饼,随后将滤饼复溶,用2n naoh调ph至碱性后,萃取、旋干,得到化合物3,即已拆分的s构型的产物2.5g白色固体,er=96.3:3.7,收率25%。
[0069]
制备化合物4:
[0070]
将得到的化合物3(2.5g,10.4mmol)溶于40ml二氯甲烷,缓慢滴加pvcl(1.4ml,11.4mol),室温反应1h;反应结束后,用饱和的氯化钠(50ml)洗涤,用乙酸乙酯(3
╳
50ml)萃取,合并有机相,用无水硫酸钠干燥,浓缩,利用硅胶柱柱层析(石油醚/乙酸乙酯),得到化合物4为无色油状(3.2g,96%);
[0071]
对化合物4进行核磁检测,得到以下数据:
[0072]1h nmr(400mhz,chloroform
‑
d)δ7.87(dd,2h),7.23(dd,2h),3.76(t,j=8.8hz,1h),3.31
–
3.24(m,3h),2.93(s,1h),1.94
–
1.55(m,4h),1.24(s,9h),
[0073]
13
c nmr(100mhz,chloroform
‑
d)δ179.5,137.5,132.0,131.8,120.6,59.1,55.4,50.9,44.1,43.8,31.9,21.5。
[0074]
制备化合物6:
[0075]
将得到的化合物4(3.2g,10.00mmol)溶于40ml dma,依次加入化合物5(2.3g,10.05mmol),溴化亚铜(72mg,0.5mmol),8
‑
羟基喹啉(145mg,1.0mmol),碳酸钾(4.1g,30.0mmol,3.0eq),氩气保护,105℃~115℃反48h;用40ml水洗,ea萃取,柱层析(pe:ea=8:1),得到化合物6白色固体(3.95g,产率83%)。
[0076]
对化合物6进行核磁检测,得到以下数据:
[0077]1h nmr(400mhz,methanol
‑
d4)δ9.54(s,1h),8.06(dd,j=7.1,1.0hz,1h),7.95
–
7.89(m,3h),7.49
–
7.42(m,2h),7.20(dd,j=8.4,7.0hz,1h),4.18
–
4.05(m,2h),2.98
–
2.68(m,3h),2.01(dd,j=11.1,3.7hz,1h),1.81
–
1.69(m,2h),1.56(s,9h),1.47(s,9h).
13
c nmr(100mhz,methanol
‑
d4)δ166.3,156.5,147.7,145.4,139.7,130.5,129.6,126.3,125.1,123.7,123.1,123.1,121.6,81.1,79.5,52.5,43.5,32.7,29.3,28.7。
[0078]
制备化合物7,即目标产物尼拉帕尼:
[0079]
将得到的化合物6(10.0g,22.0mmol)溶于40ml邻二甲苯,缓慢滴加msa(32ml,0.48mol),40℃反应3h;随后反应液中加水,分出有机相,水相用甲苯萃取弃去,取水相加饱和碳酸氢钠溶液中和,再用二氯甲烷溶液萃取水相,收集有机相浓缩干燥,将化合物溶于100ml thf,搅拌下加入对甲苯磺酸一水合物(4.00g,21mmol),65℃反应16h;过滤后,得到咖啡色固体尼拉帕尼(7.04g,产率90%);
[0080]
对得到的尼拉帕尼进行核磁检测,得到以下数据:
[0081]1h nmr(400mhz,methanol
‑
d4)δ8.97(s,1h),8.15(d,j=7.0hz,1h),8.01(t,j=8.8hz,3h),7.71(d,j=7.8hz,3h),7.49(d,j=8.2hz,2h),7.24(dd,j=17.3,7.9hz,4h),3.46(t,j=8.8hz,2h),3.18
–
3.04(m,3h),2.34(s,3h),2.07(d,j=11.7hz,2h),1.94
–
1.85(m,2h),
[0082]
13
c nmr(100mhz,methanol
‑
d4)δ169.5,147.9,142.9,141.8,140.3,131.6,129.9,129.6,127.2,127.0,125.3,124.1,123.1,122.2,121.8,50.0,45.0,40.9,30.7,23.8,21.3。
[0083]
以上所述仅为本发明的较佳实施例而已,并不用以限制本发明,凡在本发明的精神和原则之内,所作的任何修改、等同替换、改进等,均应包含在本发明的保护范围之内。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1