含色氨酸多肽烯烃化成环衍生物及其制备与应用
含色氨酸多肽烯烃化成环衍生物及其制备与应用
(一)技术领域
1.本发明涉及一种含色氨酸多肽烯烃化成环衍生物,及其制备方法和应用。
(二)
背景技术:
2.多肽是重要的生物活性分子,在药物化学、生物技术和化学生物学等领域有着广泛的应用。与线性肽相比,钉书肽具有较高的结合亲和力、靶向选择性、细胞通透性、蛋白水解稳定性和调节蛋白
‑
蛋白相互作用(ppis)的能力等突出优势。为了更好地了解它们的生理和药理功能,特别是对细胞内ppis的抑制作用,构建荧光钉书肽已被证明是药物化学领域的一种强有力的工具,因此备受关注。近年来,一些基于trp残基的后期修饰的方法已被成功开发出来。而传统对于trp残基的后期修饰的方法:通常需要先在trp的吲哚杂环的1号上引入保护基团或导向基团,完成其他位点的修饰后,最后脱保护或导向基团,实现修饰。这类方法方法步骤繁多,反应收率低,这些存在的问题大大的降低了对于含trp的多肽的后期修饰研究的进展。
(三)
技术实现要素:
3.本发明的目的是提供一种含色氨酸多肽烯烃化成环衍生物,及其制备方法和应用。
4.本发明采用的技术方案是:
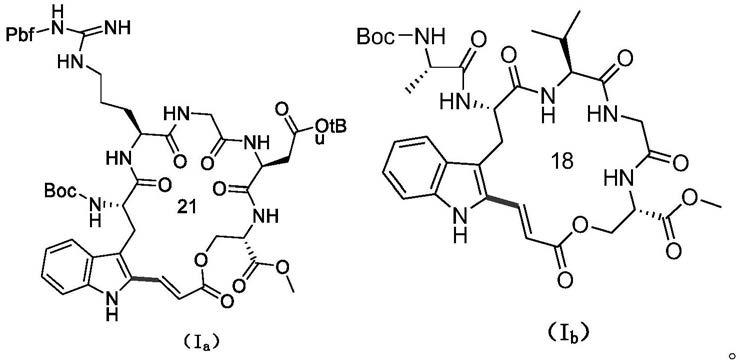
5.一种含色氨酸多肽烯烃化成环衍生物,结构如(i)所示:
[0006][0007]
式(i)中,aa为氨基酸残基,m为0~1的自然数,n为2~3的自然数。
[0008]
优选的,所述烯烃化成环衍生物为下式之一:
[0009][0010]
本发明还涉及制备式(i)所示含色氨酸多肽烯烃化成环衍生物的方法,所述方法包括:以式(ii)所示丙烯酸酯修饰的含色氨酸多肽为底物,在催化剂、氧化剂存在下,于溶剂中、在50~100℃下搅拌反应12~36小时,制得式(i)所示含色氨酸多肽烯烃化成环衍生物;
[0011][0012]
环肽前体(ii)的一般合成步骤
[0013][0014]
如图所示,将二氯树酯(300mg,0.3mmol)悬浮在5ml二氯甲烷中,然后加入fmoc
‑
aa
‑
oh(0.9mmol)和diea(154.8mg,1.2mmol),在振荡器中反应2小时后,300微升甲醇加入封端10分钟,然后用dmf将fmoc
‑
aa
‑
二氯树酯洗涤3次。fmoc
‑
aa
‑
二氯树酯用20%哌啶/dmf溶液脱fmoc保护30分钟。结束后,将h
‑
aa
‑
二氯树酯用dmf洗涤四次。随后的氨基酸偶联,使用
标准固相肽合成过程(spps),直到最后一个氨基酸boc
‑
aa
‑
oh反应结束。使用25%六氟异丙醇/二氯甲烷反应1小时把多肽从二氯树酯上裂解下来,过滤,将树酯用二氯甲烷洗涤3次,合并滤液并真空浓缩,得到多肽。最后,利用常规的液相缩合反应步骤线性肽(0.2mmol),1a(44mg,0.2mmol),edci(60mg,0.3mmol)和hobt(40mg,0.3mmol)溶解在3ml dmf中,然后加入diea(78mg,0.6mmol),在室温下搅拌12小时。反应完成后,加入10ml乙酸乙酯和10ml水,分离有机层,分别用5ml 1n盐酸,5ml饱和碳酸氢钠,5ml饱和氯化钠溶液洗涤,并用无水硫酸钠干燥,过滤,真空浓缩至得到线性肽粗品(ii),再通过硅胶柱以及制备硅胶板进一步分离纯化。
[0015]
所述催化剂为下列之一:醋酸钯,三氟乙酸钯,二氯化钯;
[0016]
所述氧化剂为下列之一:氧气,过氧苯甲酸叔丁酯,对苯醌,醋酸铜,醋酸银;
[0017]
所述溶剂为下列之一:对二甲苯,n,n
‑
二甲基甲酰胺,醋酸,甲苯,四氢呋喃/醋酸溶液,1,4二氧六环/醋酸溶液;
[0018]
所述含色氨酸多肽、催化剂、氧化剂的物质的量之比为1:0.05~0.15:0.5~2。
[0019]
具体的,所述四氢呋喃/醋酸溶液或1,4
‑
二氧六环/醋酸溶液的体积比为1~5:1。
[0020]
优选的,所述催化剂为醋酸钯,所述氧化剂为对苯醌,所述溶剂为1,4
‑
二氧六环/醋酸溶液(3:1,v/v),所述反应温度为80℃,所述反应时间为24h,所述含色氨酸多肽、催化剂、氧化剂的物质的量之比为1:0.1:1。
[0021]
所述烯烃化衍生物分离纯化方法如下:反应混合物中加入饱和nacl水溶液,用乙酸乙酯萃取,取有机层经过无水硫酸钠干燥、过滤、常温下旋转蒸除溶剂,即得粗品;将粗品进行硅胶柱层析,收集得到的洗脱液经减压除去溶剂,干燥,得到(i)所示含色氨酸多肽烯烃化成环衍生物。
[0022]
本发明还涉及所述烯烃化衍生物在制备抗肿瘤药物中的应用。
[0023]
优选的,所述肿瘤为肺癌。
[0024]
优选的,所述化合物结构如下式所示:
[0025][0026]
本发明的有益效果主要体现在:本发明化合物通过钯催化色氨酸侧链c(sp2)
‑
h直接氧化heck反应制得,操作过程简单,无导向基团,位点选择性高,反应高效,只需一步便可制得,该化合物具有较好抗肿瘤应用前景,为研发抗肿瘤药物提供了新的方案。
(四)附图说明
[0027]
图1为式(i
a
)所示化合物的核磁氢谱;
[0028]
图2为式(i
a
)所示化合物的碳谱;
[0029]
图3为式(i
b
)所示化合物的核磁氢谱;
[0030]
图4为式(i
b
)所示化合物的碳谱;
[0031]
图5为环肽化合物(i
a
)的抗肿瘤活性研究。
(五)具体实施方式
[0032]
下面结合具体实施例对本发明进行进一步描述,但本发明的保护范围并不仅限于此:
[0033]
实施例1:化合物(i
a
)的制备
[0034][0035]
如式ii
a
所示含烯烃的色氨酸线性多肽(环肽前体(ii)的一般合成步骤)(0.2mmol),对苯醌(0.4mmol),pd(oac)2(0.02mmol)称于10ml规格的圆底烧瓶中,加入10ml 1,4二氧六环/acoh=3:1溶液。盖上胶塞反应物加热至80摄氏度24小时。将40ml乙酸乙酯和20ml水加入到反应液中。有机层分别用20ml 1n盐酸,20ml饱和碳酸氢钠,20ml饱和氯化钠溶液洗涤,并用无水硫酸钠干燥,过滤,真空浓缩,得到的粗产物通过硅胶柱或制备硅胶板进一步纯化(乙酸乙酯:石油醚=4:1,r
f
=0.2),获式i
a
所示的化合物纯品80.7mg(收率40%),其核磁氢谱、碳谱参见图1~图2。
[0036]1h nmr(600mhz,dmso)δ11.43(s,1h),8.20(s,1h),8.15(d,j=8.9hz,1h),7.94(s,1h),7.89(d,j=7.2hz,1h),7.63(d,j=15.9hz,1h),7.53(d,j=8.0hz,1h),7.34(d,j=8.2hz,1h),7.22(t,j=7.5hz,1h),7.04(t,j=7.6hz,1h),6.82(d,j=9.0hz,1h),6.70(d,j=15.4hz,1h),6.40(d,j=15.9hz,1h),6.34(dd,j=17.3,1.4hz,1h),4.78
–
4.68(m,2h),4.55(d,j=8.3hz,1h),4.47
–
4.42(m,1h),4.32(dd,j=13.6,7.5hz,1h),3.79(s,1h),3.69(s,3h),3.65(d,j=6.0hz,1h),3.63(s,1h),3.29
–
3.19(m,2h),2.96(s,5h),2.84(dd,j=15.3,5.7hz,1h),2.46(s,3h),2.41(s,3h),1.41(s,8h),1.39(s,8h),1.38(s,7h).
13
c nmr(151mhz,dmso)δ172.53,171.08,169.79,169.73,169.02,166.15,157.93,156.49,155.24,138.10,137.24,133.10,131.89,131.58,129.21,124.78,120.88,120.53,119.16,116.75,114.61,86.77,80.61,79.00,62.70,60.22,56.24,52.89,49.88,42.94,37.77,28.76,28.61,28.06,27.12,21.22,19.39,18.04,14.55,12.73.ms(esi)m/z(relative intensity)1094.81(100)[m+h
+
].
[0037]
实施例2:化合物(i
b
)的制备
[0038][0039]
如式ii
b
所示含烯烃的色氨酸线性多肽(环肽前体(ii)的一般合成步骤)(0.2mmol),对苯醌(0.4mmol),pd(oac)2(0.02mmol)称于10ml规格的圆底烧瓶中,加入10ml 1,4二氧六环/acoh=3:1溶液。盖上胶塞反应物加热至80摄氏度24小时。将40ml乙酸乙酯和20ml水加入到反应液中。有机层分别用20ml 1n盐酸,20ml饱和碳酸氢钠,20ml饱和氯化钠溶液洗涤,并用无水硫酸钠干燥,过滤,真空浓缩,得到的粗产物通过硅胶柱或制备硅胶板进一步纯化(乙酸乙酯:石油醚=4:1,r
f
=0.45),获式i
b
所示的化合物纯品80.7mg(收率40%),其核磁氢谱、碳谱参见图3~图4。
[0040]1h nmr(600mhz,dmso)δ11.43(s,1h),8.20(s,1h),8.15(d,j=8.9hz,1h),7.94(s,1h),7.89(d,j=7.2hz,1h),7.63(d,j=15.9hz,1h),7.53(d,j=8.0hz,1h),7.34(d,j=8.2hz,1h),7.22(t,j=7.5hz,1h),7.04(t,j=7.6hz,1h),6.82(d,j=9.0hz,1h),6.70(d,j=15.4hz,1h),6.40(d,j=15.9hz,1h),6.34(dd,j=17.3,1.4hz,1h),4.78
–
4.68(m,2h),4.55(d,j=8.3hz,1h),4.47
–
4.42(m,1h),4.32(dd,j=13.6,7.5hz,1h),3.79(s,1h),3.69(s,3h),3.65(d,j=6.0hz,1h),3.63(s,1h),3.29
–
3.19(m,2h),2.96(s,5h),2.84(dd,j=15.3,5.7hz,1h),2.46(s,3h),2.41(s,3h),1.41(s,8h),1.39(s,8h),1.38(s,7h).
13
c nmr(151mhz,dmso)δ172.53,171.08,169.79,169.73,169.02,166.15,157.93,156.49,155.24,138.10,137.24,133.10,131.89,131.58,129.21,124.78,120.88,120.53,119.16,116.75,114.61,86.77,80.61,79.00,62.70,60.22,56.24,52.89,49.88,42.94,37.77,28.76,28.61,28.06,27.12,21.22,19.39,18.04,14.55,12.73.ms(esi)m/z(relative intensity)1094.81(100)[m+h
+
].
[0041]
实施例9:化合物(i
a
)的抗肿瘤活性检测
[0042]
选取肿瘤细胞a549(肺癌细胞),采用mtt法进行抗肿瘤细胞增殖活性检测。细胞以4000~5000个/孔的浓度接种至含有10%胎牛血清的1640培养液的96孔板中,并在板盖上加上注释,于5%co2、37℃培养12小时,待细胞在96孔板上贴壁,在无菌操作台中用移液枪加待测药物(实施例1制备的化合物(i
a
)),使每孔药物浓度分别为2μm、5μm、10μm、20μm、40μm五个浓度梯度,每个浓度设置有三个平行组,并再次将96孔板置于5%co2、37℃培养24小时。取出96孔板,向每个孔中加入10μl的mtt试剂盒试剂(购自promega公司),避光在5%co2、37℃条件下孵化4小时,吸除上清后加入150ul无菌dmso溶解甲臜,进一步在37℃培养箱中溶解5~10min,最后利用酶标仪测其吸光度。从而计算细胞活力和细胞毒性,用graphpad prism software软件处理,计算ic
50
以及ic
50 95%可信区间为22.976
±
8.221,结果见图5。
[0043]
实验结果表明,化合物(i
a
)可靶向整合素αvβ3高表达的肿瘤细胞,并具有一定的抗肿瘤活性。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1