一种氟哌利多中间体的合成方法与流程
1.本公开实施例涉及药物合成技术领域,尤其涉及一种氟哌利多中间体的合成方法。
背景技术:
2.氟哌利多用于控制急性精神病的兴奋躁动,具有神经安定作用以及增强镇痛药的镇痛作用。合成氟哌利多的关键中间体1
‑
(1,2,3,6
‑
四氢
‑4‑
吡啶基)
‑2‑
苯并咪唑啉酮,其化学结构式如下所示:
[0003][0004]1‑
(1,2,3,6
‑
四氢
‑4‑
吡啶基)
‑2‑
苯并咪唑啉酮作为合成氟哌利多的反应物,其纯度直接影响氟哌利多药物的质量以及产率。使用1
‑
(1,2,3,6
‑
四氢
‑4‑
吡啶基)
‑2‑
苯并咪唑啉酮生产氟哌利多(化合物6)的原研生产工艺为:
[0005]
在原研生产工艺中化合物6是由化合物5经pd
‑
c/氢气脱去苄基生成,其缺点在于无法有效地控制过度还原杂质化合物6
‑
1的生成,如下式:
[0006][0007]
由于该杂质(化合物6
‑
1)的物化性质和化合物6相似,很难通过常规纯化方法除去,并且其可在制备氟哌利多的步骤歩骤中生成相应的氟哌利多杂质,如下式所示:
[0008][0009]
因此这就需要在中间体6中去除杂质化合物6
‑
1,或者在氟哌利多制备的后处理中通过多次精制除去该杂质,这就无形中增加了除杂的压力,从而降低收率。
[0010]
另外,还可以脱去化合物5中的苄基,通常采用经典的氯甲酸酯法制备出化合物6的盐酸盐,如下式:
[0011][0012]
虽然这可以弥补pd
‑
c/氢气脱去苄基方法的不足,能避免还原杂质化合物6
‑
1的生成,但也存在明显的不足:氯甲酸氯甲酯具有强烈的刺鼻刺眼特性,这在工业生产中与绿色化学和环境友好理念相违背,故不适于工业化大生产。
[0013]
因此,有必要改善上述相关技术方案中存在的一个或者多个问题。
[0014]
需要注意的是,本部分旨在为权利要求书中陈述的本公开的技术方案提供背景或上下文。此处的描述不因为包括在本部分中就承认是现有技术。
技术实现要素:
[0015]
本公开实施例的目的在于提供一种氟哌利多中间体的合成方法,进而至少在一定程度上克服由于相关技术的限制和缺陷而导致的一个或者多个问题。
[0016]
本公开实施例提供一种氟哌利多中间体的合成方法,该中间体为1
‑
[1
‑
(1,2,3,6
‑
四氢
‑4‑
吡啶基)]
‑2‑
苯并咪唑啉酮盐酸盐,该合成方法包括以下步骤:
[0017]
步骤一,以1
‑
n
‑
boc
‑4‑
氧代
‑3‑
哌啶羧酸乙酯为反应物,与邻苯二胺缩合反应,得到4
‑
(2
‑
氧代
‑
2,3
‑
二氢
‑
1h
‑
苯并[d]咪唑
‑1‑
基)
‑
3,6
‑
二氢
‑
2h
‑
哌啶
‑1‑
羧酸叔丁酯;
[0018]
步骤二,把所述4
‑
(2
‑
氧代
‑
2,3
‑
二氢
‑
1h
‑
苯并[d]咪唑
‑1‑
基)
‑
3,6
‑
二氢
‑
2h
‑
哌
啶
‑1‑
羧酸叔丁酯与hcl的有机溶液反应,得到1
‑
[1
‑
(1,2,3,6
‑
四氢
‑4‑
吡啶基)]
‑2‑
苯并咪唑啉酮盐酸盐;
[0019]
上述反应的反应式如下:
[0020][0021]
本公开的一实施例中,所述步骤一的反应过程为:
[0022]
摩尔比为1:(0.8
‑
1.2)的1
‑
n
‑
boc
‑4‑
氧代
‑3‑
哌啶羧酸乙酯和邻苯二胺在第一有机溶剂中,于130
‑
150℃下反应3
‑
7h。其中,摩尔比可以为1:0.9、1:1、1:1.1等。
[0023]
本公开的一实施例中,所述第一有机溶剂为二甲苯或n
‑
甲基吡咯烷酮。
[0024]
本公开的一实施例中,所述1
‑
n
‑
boc
‑4‑
氧代
‑3‑
哌啶羧酸乙酯和邻苯二胺的反应摩尔比为1:1。
[0025]
本公开的一实施例中,将1
‑
n
‑
boc
‑4‑
氧代
‑3‑
哌啶羧酸乙酯滴加到邻苯二胺的二甲苯溶液中进行反应。
[0026]
本公开的一实施例中,所述反应在140℃下反应5h。
[0027]
本公开的一实施例中,所述步骤二的反应过程为:
[0028]
将所述步骤一制得的4
‑
(2
‑
氧代
‑
2,3
‑
二氢
‑
1h
‑
苯并[d]咪唑
‑1‑
基)
‑
3,6
‑
二氢
‑
2h
‑
哌啶
‑1‑
羧酸叔丁酯溶于第二有机溶剂中,加入1n
‑
3n的氯化氢的有机溶液,在5
‑
25℃下反应2
‑
4h。
[0029]
本公开的一实施例中,所述第二有机溶剂为二氯甲烷或乙酸乙酯。
[0030]
本公开的一实施例中,所述氯化氢的有机溶液为hcl/etoac、hcl/meoh、或hcl/et2o溶液。
[0031]
本公开的一实施例中,所述4
‑
(2
‑
氧代
‑
2,3
‑
二氢
‑
1h
‑
苯并[d]咪唑
‑1‑
基)
‑
3,6
‑
二氢
‑
2h
‑
哌啶
‑1‑
羧酸叔丁酯和氯化氢的有机溶液的反应的摩尔比为1:(1
‑
1.5)。
[0032]
本公开的实施例提供的技术方案可以包括以下有益效果:
[0033]
本公开实施例中的氟哌利多中间体的合成方法,通过采用易得化合物1
‑
n
‑
boc
‑4‑
氧代
‑3‑
哌啶羧酸乙酯为原料,研发了一种制备目标化合物的新工艺,且收率较高。该工艺不需多次精制除杂,也避免了强刺激味反应物的使用,并且易于工业化生产,解决了目前工艺中后处理纯化除杂难的问题。
附图说明
[0034]
此处的附图被并入说明书中并构成本说明书的一部分,示出了符合本公开的实施例,并与说明书一起用于解释本公开的原理。显而易见地,下面描述中的附图仅仅是本公开的一些实施例,对于本领域普通技术人员来讲,在不付出创造性劳动的前提下,还可以根据这些附图获得其他的附图。
[0035]
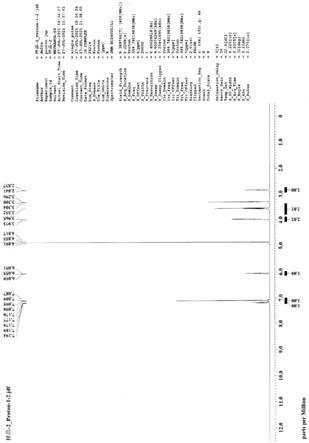
图1示出本公开示例性实施例中4
‑
(2
‑
氧代
‑
2,3
‑
二氢
‑
1h
‑
苯并[d]咪唑
‑1‑
基)
‑
3,6
‑
二氢
‑
2h
‑
哌啶
‑1‑
羧酸叔丁酯(化合物3)的核磁谱图;
[0036]
图2示出本公开示例性实施例中1
‑
[1
‑
(1,2,3,6
‑
四氢
‑4‑
吡啶基)]
‑2‑
苯并咪唑啉酮盐酸盐(化合物6盐酸盐)的核磁谱图。
具体实施方式
[0037]
现在将参考附图更全面地描述示例实施方式。然而,示例实施方式能够以多种形式实施,且不应被理解为限于在此阐述的范例;相反,提供这些实施方式使得本公开将更加全面和完整,并将示例实施方式的构思全面地传达给本领域的技术人员。所描述的特征、结构或特性可以以任何合适的方式结合在一个或更多实施方式中。
[0038]
本示例实施方式中提供一种氟哌利多中间体的合成方法,该中间体为1
‑
[1
‑
(1,2,3,6
‑
四氢
‑4‑
吡啶基)]
‑2‑
苯并咪唑啉酮盐酸盐,可以按照以下反应式进行制备:
[0039][0040]
本公开实施例中的氟哌利多中间体的合成方法,通过采用易得化合物1
‑
n
‑
boc
‑4‑
氧代
‑3‑
哌啶羧酸乙酯为原料,研发了一种制备目标化合物的新工艺,且收率较高。该工艺不需多次精制除杂,也避免了强刺激味反应物的使用,并且易于工业化生产,解决了目前工艺中后处理纯化除杂难的问题。
[0041]
下面对以上反应式的反应步骤作具体描述。
[0042]
首先,对于4
‑
(2
‑
氧代
‑
2,3
‑
二氢
‑
1h
‑
苯并[d]咪唑
‑1‑
基)
‑
3,6
‑
二氢
‑
2h
‑
哌啶
‑1‑
羧酸叔丁酯(化合物3)的制备实施例如下所述。
[0043]
实施例1:
[0044]
将邻苯二胺(27.0g)和二甲苯(300ml)加入到1l的夹套瓶中,加热至135℃。将化合物2(67.8g)预先溶于二甲苯(300ml)中,然后滴加至上述体系中,滴加完毕在此温度下反应6h。反应完毕,浓缩体系体积至约150ml,冷却至室温,滴加正己烷(500ml)至体系中,室温搅拌1
‑
2h,过滤,在45
‑
50℃烘料6h,得产品35.2g,类白色固体,收率44.7%。
[0045]
实施例2:
[0046]
将邻苯二胺(27.0g)和二甲苯(300ml)加入到1l的夹套瓶中,加热至140℃。将化合物2(67.8g)预先溶于二甲苯(300ml)中,然后滴加至上述体系中,滴加完毕在此温度下反应5h。反应完毕,浓缩体系体积至约150ml,冷却至室温,滴加正己烷(500ml)至体系中,室温搅拌1
‑
2h,过滤,在45
‑
50℃烘料6h,得产品37.6g,类白色固体,收率47.8%。
[0047]
实施例3:
[0048]
邻苯二胺(27.1g)和二甲苯(300ml)加入到1l的夹套瓶中,加热至145℃。将化合物2(67.8g)预先溶于二甲苯(300ml)中,然后滴加至上述体系中,滴加完毕在此温度下反应4h。反应完毕,浓缩体系体积至约150ml,冷却至室温,滴加正己烷(500ml)至体系中,室温搅拌1
‑
2h,过滤,在45
‑
50℃烘料6h,得产品34.8g,类白色固体,收率44.2%。
[0049]
其次,1
‑
(1,2,3,6
‑
四氢
‑4‑
吡啶基)
‑2‑
苯并咪唑啉酮盐酸盐(化合物6盐酸盐)的制备实施例如下所述。
[0050]
实施例4:
[0051]
将化合物3(50.0g)和二氯甲烷(250ml)加入到500ml的夹套瓶中,在25℃下滴加4n hcl/etoac溶液(50ml),滴加完毕,搅拌2h。反应完毕,过滤,滤饼用二氯甲烷(100ml)洗涤,在45
‑
50℃烘料6h,得产品38.2g,类白色固体,收率95.7%。
[0052]
实施例5:
[0053]
将化合物3(50.0g)和二氯甲烷(250ml)加入到500ml的夹套瓶中,在15℃滴加4n hcl/etoac溶液(50ml),滴加完毕,室温下搅拌4h。反应完毕,过滤,滤饼用二氯甲烷(100ml)洗涤,在45
‑
50℃烘料6h,得产品38.6g,类白色固体,收率96.6%。
[0054]
实施例6:
[0055]
将化合物3(50.0g)和二氯甲烷(200ml)加入到500ml的夹套瓶中,在5℃滴加4n hcl/etoac溶液(50ml),滴加完毕,在15℃搅拌4h。反应完毕,过滤,滤饼用二氯甲烷(80ml)洗涤,在45
‑
50℃烘料6h,得产品39.0g,类白色固体,收率97.7%。
[0056]
其中,化合物6盐酸盐制备过程中使用的氯化氢的盐酸盐溶液可以为hcl/etoac(乙酸乙酯)、hcl/meoh(甲醇)、或hcl/et2o(乙醚)溶液。所述4
‑
(2
‑
氧代
‑
2,3
‑
二氢
‑
1h
‑
苯并[d]咪唑
‑1‑
基)
‑
3,6
‑
二氢
‑
2h
‑
哌啶
‑1‑
羧酸叔丁酯和氯化氢的有机溶液的反应的摩尔比为1:(1
‑
1.5),例如可以是1:1.2、1:1.26、1:1.3等,其中1:1.26最佳。
[0057]
对以上实施例制备出的化合物3、化合物6盐酸盐分别进行了核磁测试和表征,分别得到图1、图2的核磁谱图。由图1可以得到:1h nmr(400mhz,d6
‑
dmso):10.960(1h,s,nh),7.068
‑
6.989(4h,m,arh),5.954
‑
5.920(1h,m,c=ch),4.068
‑
4.063(2h,m,ch2),3.611
‑
3.583(2h,m,ch2),2.498
‑
2.494(2h,m ch2),1.286(9h,s,3ch3)。由图2可以得到:1h nmr(400mhz,d
‑
meod):7.194
‑
7.170(1h,m,arh),7.098
‑
7.087(3h,m,arh),6.059
‑
6.051(1h,m,c=ch),3.973
‑
3.965(2h,m,ch2),3.558
‑
3.551(2h,m ch2),2.841
‑
3.2.837(2h,m ch2)。验证了化合物3、化合物6盐酸盐的结构式分别如下:
[0058][0059]
通过本发明的方法制备出的1
‑
[1
‑
(1,2,3,6
‑
四氢
‑4‑
吡啶基)]
‑2‑
苯并咪唑啉酮盐酸盐可以直接用于生产氟哌利多的生产,有利于提高氟哌利多的合成质量以及产率。
[0060]
需要理解的是,术语“第一”、“第二”仅用于描述目的,而不能理解为指示或暗示相对重要性或者隐含指明所指示的技术特征的数量。由此,限定有“第一”、“第二”的特征可以明示或者隐含地包括一个或者更多个该特征。在本公开实施例的描述中,“多个”的含义是两个或两个以上,除非另有明确具体的限定。
[0061]
在本说明书的描述中,参考术语“一个实施例”、“一些实施例”、“示例”、“具体示例”、或“一些示例”等的描述意指结合该实施例或示例描述的具体特征、结构、材料或者特点包含于本公开的至少一个实施例或示例中。在本说明书中,对上述术语的示意性表述不必须针对的是相同的实施例或示例。而且,描述的具体特征、结构、材料或者特点可以在任何的一个或多个实施例或示例中以合适的方式结合。此外,本领域的技术人员可以将本说明书中描述的不同实施例或示例进行接合和组合。
[0062]
本领域技术人员在考虑说明书及实践这里公开的发明后,将容易想到本公开的其它实施方案。本技术旨在涵盖本公开的任何变型、用途或者适应性变化,这些变型、用途或者适应性变化遵循本公开的一般性原理并包括本公开未公开的本技术领域中的公知常识或惯用技术手段。说明书和实施例仅被视为示例性的,本公开的真正范围和精神由所附的权利要求指出。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1