一种2,2-双(4-氨基苯基)六氟丙烷的绿色制备方法与流程
一种2,2
‑
双(4
‑
氨基苯基)六氟丙烷的绿色制备方法
技术领域
1.本发明涉及精细有机合成技术领域,尤其涉及一种2,2
‑
双(4
‑
氨基苯基)六氟丙烷的绿色制备方法。
背景技术:
2.聚酰亚胺作为金字塔顶端的特种工程塑料,具有优异的热性能、物理机械性能和化学稳定性,适用于各领域。在聚酰亚胺中引入氟的含氟聚酰亚胺有上述特性之外,兼具高分子材料特点,这些性能使其在电子材料、航空航天用材料、光学材料、分离膜、液晶材料等领域具有重要的应用前景。通常情况,含氟聚酰亚胺是由含氟二胺化合物与芳香性二酐化合物反应制备而成,或由含氟芳香性二酐化合物与二胺化合物反应制备而成。
3.在各种含氟聚酰亚胺中间体中,2,2
‑
双(4
‑
氨基苯基)六氟丙烷是一种被广泛选用的中间体。目前,公开报道的2,2
‑
双(4
‑
氨基苯基)六氟丙烷的合成方法主要有两种。其中一种是maruta研究团队提出的三步合成法,即a)2,2
‑
双(4
‑
羧酸苯基)
‑
六氟丙烷为原料与二氯亚砜反应生成2,2
‑
双(4
‑
氯甲酰基苯基)
‑
六氟丙烷;b)由2,2
‑
双(4
‑
氯甲酰基苯基)
‑
六氟丙烷与叠氮化合物反应生成2,2
‑
双(4
‑
异氰酸酯基苯基)
‑
六氟丙烷;c)2,2
‑
双(4
‑
异氰酸酯基苯基)
‑
六氟丙烷的异氰酸酯基团水解得2,2
‑
双(4
‑
氨基苯基)
‑
六氟丙烷。另一种合成方法是由美国专利公开号为us4370501a公开,首先以2,2
‑
双(4
‑
羟基苯基)
‑
六氟丙烷钾盐为原料,在二甲基亚砜条件下将其转换为4
‑
氯
‑2‑
苯基喹唑啉,然后在320℃高温下将4
‑
氯
‑2‑
苯基喹唑啉转变为双喹唑啉酮,最后再将双喹唑啉酮水解成2,2
‑
双(4
‑
氨基苯基)
‑
六氟丙烷。基于上述两种方法,前一种方法使用叠氮化合物存在操作危险性较高,同时形成的异氰酸中间化合物难以分离的问题;后一种方法所需试剂价格昂贵,反应条件比较严苛,不适合工业化生产。
4.另有,专利公开号为cn108358794a公开了一种2,2
‑
双(4
‑
氨基苯基)
‑
六氟丙烷的合成新方法,其使用的重金属氧化剂,氧化剂成本高,并存在严重的环境污染和职业健康风险,并且产品中残留重金属的繁杂去除工艺会增加生产成本,不适合工业化生产。
5.综上所述,现有的2,2
‑
双(4
‑
氨基苯基)
‑
六氟丙烷合成方法均存在一些技术问题,开发一种新的、安全性高、成本低的合成方法显得十分紧迫和必要。
技术实现要素:
6.针对现有技术的不足,本发明的目的在于克服现有2,2
‑
双(4
‑
氨基苯基)六氟丙烷制备方法存在的重金属氧化剂昂贵、溶剂易燃易爆危险性高、反应条件苛刻等问题,提供一种2,2
‑
双(4
‑
氨基苯基)六氟丙烷的绿色的合成方法。
7.本发明采用以下技术方案:
8.一种2,2
‑
双(4
‑
氨基苯基)六氟丙烷的绿色制备方法,包括以下步骤:
9.(a)在全氟化合物溶剂中,2,2
‑
双(4
‑
甲基苯基)六氟丙烷和氧气发生氧化反应,分离得到2,2
‑
双(4
‑
羧基苯基)六氟丙烷;
10.(b)将步骤(a)得到的化合物与氨气在有机溶剂中回流反应,得到2,2
‑
双(4
‑
羧基苯基)六氟丙烷的铵盐;
11.(c)将步骤(b)得到的化合物与有机溶剂混合加热,升温脱水,得到2,2
‑
双(4
‑
甲酰胺基苯基)六氟丙烷;
12.(d)将步骤(c)得到的化合物与氢氟醚配制成溶液,滴加入次氯酸钠进行霍夫曼降解反应,纯化得到产物2,2
‑
双(4
‑
氨基苯基)六氟丙烷。
13.进一步的,步骤(a)中,2,2
‑
双(4
‑
甲基苯基)六氟丙烷与全氟化合物溶剂的质量比为1:(2
‑
6),2,2
‑
双(4
‑
甲基苯基)六氟丙烷与氧气的摩尔比为1:(3
‑
5),反应温度90
‑
150℃,反应时间12
‑
16h,氧气从反应器的液相鼓泡通入。选用恰当的2,2
‑
双(4
‑
甲基苯基)六氟丙烷与全氟化合物溶剂的质量比,可完全溶解反应物2,2
‑
双(4
‑
甲基苯基)六氟丙烷。全氟化合物溶剂用量过少时,反应物会出现不完全溶解现象;全氟化合物溶剂用量过高时,系统利用率低,产能小。
14.优选的,步骤(a)为:将2,2
‑
双(4
‑
甲基苯基)六氟丙烷加入到全氟化合物溶剂中,搅拌并升温至90
‑
120℃,待固体完全溶解后,连续鼓泡从反应器的液相通入氧气,控制反应内温为90
‑
150℃反应12
‑
16h,降温,析出,过滤,得到中间产物2,2
‑
双(4
‑
羧基苯基)六氟丙烷。
15.进一步的,所述全氟化合物溶剂为全氟烷烃、全氟环烷烃、全氟二烷基醚、全氟聚醚、全氟取代的芳香族化合物、全氟胺类化合物中的一种。现有技术中,制备中间产物2,2
‑
双(4
‑
羧基苯基)六氟丙烷时反应溶剂多为乙酸或乙酸酐,氧化剂为氧气,同时添加催化剂。此过程中,存在燃爆风险;催化剂消耗量大,2,2
‑
双(4
‑
甲基苯基)六氟丙烷氧化不完全,需要过量的氧气,考虑到工业生产安全,需要开发不易燃,不需要添加催化剂的制备方法。本发明选用全氟化合物溶剂,全氟化合物溶剂具有抗氧化、不燃不爆的特性;其作为氧化反应的安全溶剂,可循环利用,有利于安全生产。本发明中全氟化合物溶剂溶解氧气程度高,可以直接与氧气接触,不燃,反应过程安全稳定,不需要额外添加催化剂,适合工业化生产。
16.优选的,步骤(a)中,所述全氟化合物溶剂为氟碳环醚、全氟(2
‑
正丁基四氢吡喃)、全氟聚醚中的一种。
17.进一步的,步骤(b)中,2,2
‑
双(4
‑
羧基苯基)六氟丙烷与氨气的摩尔比为1:(2
‑
2.5)。优选的,步骤(b)中,2,2
‑
双(4
‑
羧基苯基)六氟丙烷与氨气的摩尔比为1:(2.2
‑
2.5)。
18.进一步的,步骤(b)、(c)中,所述有机溶剂为芳香烃化合物、全氟化合物,氢氟醚化合物中的一种;步骤(c)中,升温脱水的温度为120
‑
200℃。优选的,步骤(b)中,所述芳香烃化合物为二甲苯、邻二甲苯、对二甲苯、三甲苯、萘中的一种。在2,2
‑
双(4
‑
甲酰胺基苯基)六氟丙烷的制备过程中,无需使用氯化试剂,工艺简单,制造成本低。
19.进一步的,步骤(d)中,将2,2
‑
双(4
‑
甲酰胺基苯基)六氟丙烷与氢氟醚配制成溶液,降温,滴加入次氯酸钠溶液,升温后,滴加氢氧化钠溶液,分层,取下层溶液,经浓缩、烘干,纯化得到2,2
‑
双(4
‑
氨基苯基)六氟丙烷。优选的,步骤(d)中,将2,2
‑
双(4
‑
甲酰胺基苯基)六氟丙烷溶解在氢氟醚溶剂中,降温至0
‑
10℃。向降温至0℃的次氯酸钠溶液中滴加溶有2,2
‑
双(4
‑
甲酰胺基苯基)六氟丙烷的氢氟醚溶剂,搅拌1
‑
2h,升温至60℃,滴加氢氧化钠溶液后,维持搅拌0.5
‑
2h,取反应液测ph值,反应终点以所测ph值为碱性为准。确认反应完后分层,取下层溶液经浓缩、烘干,得到2,2
‑
双(4
‑
氨基苯基)六氟丙烷。
20.进一步的,步骤(d)中,所述氢氟醚为全氟丁基甲醚、3
‑
甲氧基
‑2‑
三氟甲基八氟丁烷、十氟
‑3‑
甲氧基
‑2‑
三氟甲基戊烷中的一种或多种。
21.本发明的有益效果主要体现为:
22.本发明制备的2,2
‑
双(4
‑
氨基苯基)六氟丙烷选用不燃不爆的全氟化合物溶剂,同时采用氧气做氧化剂,全氟化合物溶剂溶解氧气程度高,可以直接与氧气接触,作为氧化反应的安全溶剂,可循环利用,有利于安全生产;在2,2
‑
双(4
‑
甲酰胺基苯基)六氟丙烷的制备过程中,无需使用氯化试剂,工艺简单,制造成本低;在制备2,2
‑
双(4
‑
氨基苯基)六氟丙烷的最后阶段,使用氢氟醚,取代醇溶剂,氢氟醚对2,2
‑
双(4
‑
氨基苯基)六氟丙烷溶解度较好,并且能与水较好分层,不仅节省了萃取工序,同时上层水体系中不含醇溶剂,废水cod低,大大减轻了废水的处理成本,再者,氢氟醚通过旋蒸烘干实现回收利用,有效地节约了大量溶剂的成本,有利于高效绿色工业化生产。本发明的2,2
‑
双(4
‑
氨基苯基)六氟丙烷的绿色制备方法,选用绿色经济的氧气氧化剂,不燃不爆的全氟溶剂作为氧化溶剂,使用氢氟醚作为霍夫曼反应溶剂,有效减少高cod废水,反应速度快,反应收率高,原料廉价易得,反应条件温和,提升了工艺安全性,降低了工艺成本,容易工业化大规模生产。
具体实施方式
23.下面将对本发明实施例中的技术方案进行清楚、完整的描述,显然,所描述的实施例仅仅是本发明一部分实施例,而不是全部的实施例。基于本发明中的实施例,本领域普通的技术人员在没有做出创造性劳动的前提下所获得的所有其它实施例,都属于本发明的保护范围。
24.一种2,2
‑
双(4
‑
氨基苯基)六氟丙烷的绿色制备方法,包括以下步骤:
25.(a)将2,2
‑
双(4
‑
甲基苯基)六氟丙烷加入到全氟化合物溶剂中,搅拌并升温至90
‑
120℃,待固体完全溶解后连续通入氧气,控制反应内温为90
‑
150℃反应12
‑
16h,最后降温,析出,过滤即得中间产物2,2
‑
双(4
‑
羧基苯基)六氟丙烷;
26.(b)将2,2
‑
双(4
‑
羧基苯基)六氟丙烷与氨气在有机溶剂中回流反应,得到2,2
‑
双(4
‑
羧基苯基)六氟丙烷的铵盐,2,2
‑
双(4羧基苯基)六氟丙烷与氨气的摩尔比为1:(2
‑
2.5);
27.(c)将2,2
‑
双(4
‑
羧基苯基)六氟丙烷的铵盐与有机溶剂混合加热,升温脱水,温度为120
‑
200℃,得到2,2
‑
双(4
‑
甲酰胺基苯基)六氟丙烷;
28.(d)将2,2
‑
双(4
‑
甲酰胺基苯基)六氟丙烷与氢氟醚配制成溶液,降温至0
‑
10℃。向降温至0℃的次氯酸钠溶液中滴加溶有2,2
‑
双(4
‑
甲酰胺基苯基)六氟丙烷的氢氟醚溶液,搅拌1
‑
2h,升温至60℃,滴加氢氧化钠溶液后,继续搅拌0.5
‑
2h,取反应液测ph值,反应终点以所测ph值为碱性为准。确认反应完后分层,取下层溶液经浓缩、烘干,得到2,2
‑
双(4
‑
氨基苯基)六氟丙烷。
29.本发明的具体反应过程,如下所示:
30.步骤(a)反应式如下:
31.32.步骤(b、c)反应式如下:
[0033][0034]
步骤(d)反应式如下:
[0035][0036]
本发明所使用的原料如无特殊说明均为普通市售,ar纯。
[0037]
实施例1
[0038]
一种2,2
‑
双(4
‑
氨基苯基)六氟丙烷的绿色制备方法,包括以下步骤:
[0039]
(a)2,2
‑
双(4
‑
羧基苯基)六氟丙烷的合成
[0040]
称取3320g(10mol)2,2
‑
双(4
‑
甲基苯基)六氟丙烷、9960g氟碳环醚溶剂加入到10升(φ10cm
×
130cm)的不锈钢管式反应器中边搅拌边升温至90℃使固体溶解。待固体全部溶解后,经液相分布器通入739l(标准大气压下)氧气(33mol),控制反应温度维持在90
‑
120℃。反应完毕后,降温至室温,固体析出、过滤,所得固体在60℃下真空干燥,得到白色晶体状固体产物3737g。以2,2
‑
双(4
‑
甲基苯基)六氟丙烷计,摩尔收率为95.3%。对产物进行h nmr(400mhz,dmso)分析表征,结果如下:13.32(s,2h),8.08(d,j=8hz,4h),7.51(d,j=8hz,4h),说明该产物的确是2,2
‑
双(4
‑
羧基苯基)六氟丙烷,纯度为99.5%。
[0041]
(b)2,2
‑
双(4
‑
羧基苯基)六氟丙烷的铵盐的合成
[0042]
称取(a)制备得到的2,2
‑
双(4
‑
羧基苯基)六氟丙烷90g(0.23mol)置于三口烧瓶中,加入400ml二氯甲烷加热至回流使固体溶解(固体完全溶解,溶液清澈),通入11.8l(0.53mol,标准大气压下)氨气,得2,2
‑
双(4
‑
羧基苯基)六氟丙烷的铵盐。
[0043]
(c)2,2
‑
双(4
‑
甲酰胺基苯基)六氟丙烷化合物的合成
[0044]
将(b)制备得到的2,2
‑
双(4
‑
羧基苯基)六氟丙烷的铵盐与邻二甲苯混合加热,升温至回流温度下,内温为150
‑
170℃,脱水4小时。通过取样进行hplc测试确认脱水完全后,冷却析出得到2,2
‑
双(4
‑
甲酰胺基苯基)六氟丙烷,经hplc测试含量为99.7%。
[0045]
(d)2,2
‑
双(4
‑
氨基苯基)六氟丙烷的合成
[0046]
取1升带有回流冷凝管的三颈烧瓶加入137.5g(0.24mol)质量分数为13%的次氯酸钠,并冷却至0℃。将23.4g(0.06mol)步骤(c)中得到的2,2
‑
双(4
‑
甲酰胺基苯基)六氟丙烷与93.6g全氟丁基甲醚混合配制成溶液,将该溶液逐渐滴加到三颈烧瓶中,且三颈烧瓶内部温度保持在不高于5℃。加入完成之后,所得混合物在不高于5℃的温度下搅拌1小时。将所得反应溶液加入200g水稀释,加热至60
‑
62℃回流并搅拌1小时,冷却至20℃。冷却完成后,加入25%的氢氧化钠溶液使反应溶液的ph值为9,通过分液漏斗分层,得到下层溶液为2,2
‑
双(4
‑
氨基苯基)六氟丙烷的全氟丁基甲醚溶液。将下层溶液通过旋蒸仪烘干回收全氟丁基甲醚,得到固体为目标化合物2,2
‑
双(4
‑
氨基苯基)六氟丙烷,经hplc测试,纯度为99.83%。
[0047]
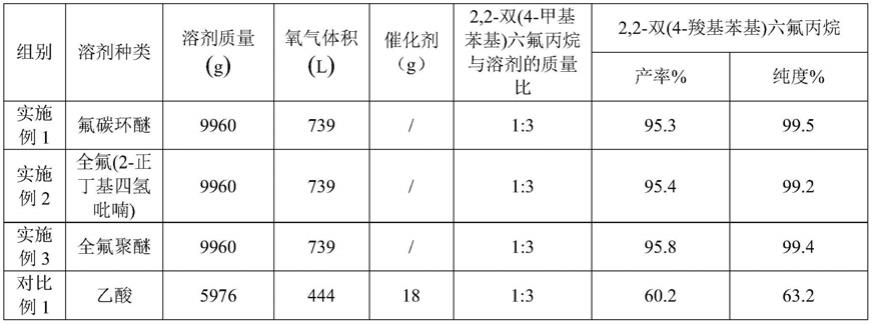
实施例1步骤(a)产物产率、纯度数据见表1。
[0048]
实施例2
[0049]
如表1所示,除了将实施例1步骤(a)中溶剂换成全氟(2
‑
正丁基四氢吡喃)之外,其
他与实施例1相同。步骤(a)产物产率、纯度数据见表1。
[0050]
实施例3
[0051]
如表1所示,除了将实施例1步骤(a)中溶剂换成全氟聚醚之外,其他与实施例1相同。步骤(a)产物产率、纯度数据见表1。
[0052]
对比例1
[0053]
2,2
‑
双(4
‑
羧基苯基)六氟丙烷的合成
[0054]
称取1992g(6mol)2,2
‑
双(4
‑
甲基苯基)六氟丙烷、5976g乙酸加入到10升(φ10cm
×
130cm)的不锈钢管式反应器,接着加入催化剂12g溴化钴、6g醋酸锰,搅拌使形成均一溶液。升温至120℃后,经液相分布器通入444l(标准大气压下)氧气(19.8mol),控制反应温度维持在90
‑
120℃。反应完毕后,经水洗、萃取、烘干,所得固体在60℃下真空干燥,得到2,2
‑
双(4
‑
羧基苯基)六氟丙烷。产率、纯度数据见表1。
[0055]
表1
[0056][0057]
根据实施例1
‑
3与对比例1测试结果可知,本发明步骤(a)中选用全氟化合物溶剂,实施例1
‑
3通过添加不同种类的全氟化合物溶剂,且没有额外添加催化剂的情况下,均能够成功合成2,2
‑
双(4
‑
羧基苯基)六氟丙烷,同时其收率和纯度均较高,且最终得到的2,2
‑
双(4
‑
氨基苯基)六氟丙烷也具有较高的产率和纯度。本发明不同种类的全氟化合物溶剂溶解氧气程度高,可以直接与氧气接触,不燃,反应过程安全稳定。对比例1选用乙酸替换氟碳环醚,只有在同时添加催化剂的情况下才能反映,且对比例1制备得到的2,2
‑
双(4
‑
羧基苯基)六氟丙烷产率、纯度与实施例1
‑
3均相差较大。
[0058]
实施例4
[0059]
2,2
‑
双(4
‑
羧基苯基)六氟丙烷的合成
[0060]
称取1660g(5mol)2,2
‑
双(4
‑
甲基苯基)六氟丙烷、4980g氟碳环醚溶剂加入到10升(φ10cm
×
130cm)的不锈钢管式反应器中边搅拌边升温至90℃使固体溶解。待固体全部溶解后,经液相分布器通入370升(标准大气压下)氧气(16.5mol),控制反应温度维持在90
‑
120℃。反应完毕后,降温至室温,固体析出、过滤,所得固体在60℃下真空干燥,得到产物2,2
‑
双(4
‑
羧基苯基)六氟丙烷。对产物进行h nmr(400mhz,dmso)分析表征,结果如下:13.32(s,2h),8.08(d,j=8hz,4h),7.51(d,j=8hz,4h),说明该产物的确是2,2
‑
双(4
‑
羧基苯基)六氟丙烷。产物产率、纯度数据见表2。
[0061]
实施例5
[0062]
如表2所示,除了将实施例4中氟碳环醚的投料量换成3320g之外,其他条件与实施
例4相同。产物产率、纯度数据见表2。
[0063]
实施例6
[0064]
如表2所示,除了将实施例4中氟碳环醚的投料量换成6640g之外,其他条件与实施例4相同。产物产率、纯度数据见表2。
[0065]
实施例7
[0066]
如表2所示,除了将实施例4中氟碳环醚的投料量换成8300g之外,其他条件与实施例4相同。产物产率、纯度数据见表2。
[0067]
对比例2
[0068]
如表2所示,除了将实施例4中氟碳环醚的投料量换成2490g之外,其他条件与实施例4相同。产物产率、纯度数据见表2。
[0069]
对比例3
[0070]
如表2所示,除了将实施例4中氟碳环醚的投料量换成11620g之外,其他条件与实施例4相同。产物产率、纯度数据见表2。
[0071]
表2
[0072][0073]
根据实施例4
‑
7与对比例2
‑
3测试结果可知,本发明实施例4
‑
7中,2,2
‑
双(4
‑
甲基苯基)六氟丙烷与全氟化合物溶剂的用量在1:(2
‑
6)范围内,均成功合成了2,2
‑
双(4
‑
羧基苯基)六氟丙烷,同时其产率和纯度均较高。当全氟化合物溶剂用量少时,如对比例2所示,反应物会出现不完全溶解现象,使得得到的产率及纯度均较低;当全氟化合物溶剂用量过高时,如对比例3所示,得到的2,2
‑
双(4
‑
羧基苯基)六氟丙烷的产率和纯度虽差距不大,但存在溶剂过量、系统利用率低的问题,产能小。
[0074]
实施例8
[0075]
如表3所示,除了将实施例1中氧气的体积换成806l之外,其他条件与实施例1相同。步骤(a)产物产率、纯度数据见表3。
[0076]
实施例9
[0077]
如表3所示,除了将实施例1中氧气的体积换成896l之外,其他条件与实施例1相同。步骤(a)产物产率、纯度数据见表3。
[0078]
对比例4
[0079]
如表3所示,除了将实施例1中氧气的体积换成448l之外,其他条件与实施例1相同。步骤(a)产物产率、纯度数据见表3。
[0080]
表3
[0081][0082]
根据实施例1、实施例8
‑
9与对比例4测试结果可知,实施例1、实施例8
‑
9通过改变氧化剂氧气的用量,控制2,2
‑
双(4
‑
甲基苯基)六氟丙烷与氧气的摩尔比为1:(3
‑
5),均成功合成了2,2
‑
双(4
‑
羧基苯基)六氟丙烷,同时其收率和纯度均较高。当氧气含量较低时,如对比例4所示,得到的2,2
‑
双(4
‑
羧基苯基)六氟丙烷纯度及收率相较于实施例1偏低。当氧气含量过高时,过量的氧气增加反应成本,且对产物的产率和纯度提升作用不大。本发明选用氧气作为氧化剂,不仅省去重金属氧化剂成本,也解决了重金属带来的环保问题,适合工业生产。
[0083]
综上,本发明制备的2,2
‑
双(4
‑
氨基苯基)六氟丙烷选用全氟化合物溶剂,作为氧化反应的安全溶剂,可循环利用,有利于安全生产,制备得到较高纯度和收率的2,2
‑
双(4
‑
羧基苯基)六氟丙烷;在2,2
‑
双(4
‑
甲酰胺基苯基)六氟丙烷的制备过程中,无需使用氯化试剂,工艺简单,制造成本低;采用以氢氟醚作为原料的溶剂进行霍夫曼降解反应得到2,2
‑
双(4
‑
氨基苯基)六氟丙烷,可大大减少废水中cod的产生,同时可以实现溶剂循环利用,节约成本,有助于绿色生产。本发明的2,2
‑
双(4
‑
氨基苯基)六氟丙烷的绿色制备方法,原料廉价易得,反应条件温和,容易工业化大规模生产。
[0084]
以上借助具体实施例对本发明做了进一步描述,但是应该理解的是,这里具体的描述,不应理解为对本发明的实质和范围的限定,本领域内的普通技术人员在阅读本说明书后对上述实施例做出的各种修改,都属于本发明所保护的范围。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1