用于递送核酸的新型脂质和脂质纳米颗粒制剂的制作方法
用于递送核酸的新型脂质和脂质纳米颗粒制剂
1.本技术是2016年10月28日提交的发明名称为“用于递送核酸的新型脂质和脂质纳米颗粒制剂”的第201680063235.2号中国专利申请的分案申请。
2.背景
技术领域
3.本发明总体上涉及新型阳离子脂质,其可用于与其他脂质组分(例如中性脂质、胆固醇和聚合物缀合的脂质)结合,以便形成具有寡核苷酸的脂质纳米颗粒,从而促进体外和体内的治疗性核酸(例如,寡核苷酸、信使rna)的细胞内递送。
4.相关领域描述
5.关于递送核酸以便在生物系统中影响所期望的应答,还存在着诸多挑战。基于核酸的治疗具有巨大潜力,但需要将核酸更有效地递送至细胞或生物体内的适当位点,以便实现这种潜力。治疗性核酸包括,例如,信使rna(mrna)、反义寡核苷酸、核酶、dna酶、质粒、免疫刺激核酸、antagomir、antimir、模拟物(mimic)、supermir和适配子。一些核酸,如mrna或质粒,可以用于实现特定细胞产物的表达,正如其对于治疗例如与蛋白质或酶的缺乏有关的疾病是有用的。可翻译的核苷酸递送的治疗性应用是极其广泛的,这是因为可以合成构建体以产生任何所选蛋白质序列(不论是否为该系统固有的)。所述核酸的表达产物可以提高蛋白质的存在水平,取代缺失的或非功能的蛋白质形式,或在细胞或生物体内引入新的蛋白质以及相关功能。
6.一些核酸,如mirna抑制剂,可以用于实现由mirna调控的特定细胞产物的表达,正如其对于治疗例如与蛋白质或酶的缺乏有关的疾病是有用的。mirna抑制的治疗性应用是极其广泛的,这是因为可以合成构建体以抑制一种或多种进而调控mrna产物表达的mirna。内源性mirna的抑制可以增加其下游的靶内源性蛋白质表达,并且恢复细胞或生物体内的适当功能,作为治疗与特定mirna或mirna组有关的疾病的手段。
7.其他的核酸可以下调特定mrna的细胞内水平,并且,结果是,通过诸如rna干扰(rnai)或反义rna的互补结合的过程下调相应的蛋白质的合成。反义寡核苷酸和rnai的治疗性应用也是极其广泛的,因为可以用针对靶mrna指导的任何核苷酸序列合成寡核苷酸构建体。靶标可以包括来自正常细胞的mrna、与病症(例如癌症)有关的mrna以及感染因子(例如病毒)的mrna。迄今为止,反义寡核苷酸构建体已显示出在体外和体内模型中通过同源mrna的降解来特异性地下调靶蛋白质的能力。另外,反义寡核苷酸构建体目前正在临床研究中被评估。
8.然而,在治疗环境中使用寡核苷酸目前面临着两个问题。第一,游离的rna易于在血浆中核酸酶消化。第二,游离rna进入存在相关翻译机制的细胞内隔室的能力受限。由阳离子脂质与其他脂质组分(如中性脂质、胆固醇、peg、peg化的脂质和寡核苷酸)形成的脂质纳米颗粒已用于阻止rna在血浆中的降解并促进寡核苷酸的细胞摄取。
9.仍然需要改进的用于递送寡核苷酸的阳离子脂质和脂质纳米颗粒。优选地,这些脂质纳米颗粒会提供优化的药物:脂质比,保护核酸不在血清中被降解和清除,其适于全身
或局部递送,并且提供核酸的细胞内递送。另外,这些脂质
‑
核酸颗粒应当是耐受良好的,并且提供足够的治疗指数,使得在有效剂量的核酸下的患者治疗与对患者产生不可接受的毒性和/或风险无关。本发明提供这些优点和相关的优点。
10.概述
11.简言之,本发明提供了脂质化合物,包括其立体异构体、药物可接受的盐或互变异构体,其可以单独使用,或者与其他脂质组分例如中性脂质、带电脂质、类固醇(包括例如,所有固醇类)和/或它们的类似物、和/或聚合物缀合的脂质联合使用,以形成用于递送治疗剂的脂质纳米颗粒。在一些实例中,将脂质纳米颗粒用于递送核酸,例如反义rna和/或信使rna。还提供了使用这类脂质纳米颗粒来治疗各种疾病或病况(如感染性实体和/或蛋白质缺乏引起的疾病或病况)的方法。
12.在一个实施方案中,提供了具有以下结构(i)的化合物:
[0013][0014]
或其药物可接受的盐、互变异构体或立体异构体,其中r1、r2、r3、l1、l2、g1、g2和g3如本文所定义。
[0015]
还提供了包含一种或多种前述结构(i)的化合物和治疗剂的药物组合物。在一些实施方案中,药物组合物还包含选自中性脂质、带电脂质、类固醇和聚合物缀合的脂质的一种或多种组分。这类组合物对于形成用于递送治疗剂的脂质纳米颗粒是有用的。
[0016]
在其他实施方案中,本发明提供了向有需要的患者施用治疗剂的方法,该方法包括制备包含结构(i)的化合物的脂质纳米颗粒和治疗剂的组合物,并向患者递送组合物。
[0017]
本发明的这些和其他方面经参考以下详细描述将会是显而易见的。
[0018]
附图中几个视图的简述
[0019]
在附图中,相同的参考编号表示类似的要素。图中要素的尺寸和相对位置不一定是按比例绘制的,并且这些要素中的一些被任意地放大和放置以改善图的易读性。而且,所绘制要素的特定形状并非旨在表达有关特定要素的实际形状的信息,并且仅选择以便易于图中的识别。图1示出了鼠肝脏中荧光素酶表达的时间过程。
[0020]
图2例示了对于作为与所公开脂质相关的代表性实例的mc3的pka的计算。
[0021]
图3提供了选定的脂质的比较性荧光素酶活性数据。
[0022]
发明详述
[0023]
在以下描述中,阐述了某些具体的详细内容,以便提供对本发明各种实施方案的透彻理解。然而,本领域技术人员将理解,本发明可以在没有这些详细内容的情况下实施。
[0024]
本发明部分基于新型阳离子(氨基)脂质的发现,当所述脂质在用于将活性剂或治疗剂(例如核酸)体内递送至哺乳动物细胞中的脂质纳米颗粒中使用时,其提供了优点。特别地,本发明的实施方案提供了包含一种或多种本文所述的新型的阳离子脂质的核酸
‑
脂质纳米颗粒组合物,其提供增强的核酸活性和改善的组合物体内耐受性,使得与先前描述的核酸
‑
脂质纳米颗粒组合物相比,治疗指数显著提高。
[0025]
在特定的实施方案中,本发明提供了新型的阳离子脂质,其能够配制用于体外和体内递送mrna和/或其他寡核苷酸的改进的组合物。在一些实施方案中,这些改进的脂质纳米颗粒组合物用于表达由mrna编码的蛋白质。在其他实施方案中,这些改进的脂质纳米颗粒组合物通过递送靶向一种特定mirna或一组mirna(其调节一种靶mrna或多种mrna)的mirna抑制剂从而用于上调内源性蛋白质表达。在其他实施方案中,这些改进的脂质纳米颗粒组合物用于下调(例如,沉默)靶基因的蛋白质水平和/或mrna水平。在一些其他实施方案中,脂质纳米颗粒还用于递送用来转基因表达的mrna和质粒。在其他实施方案中,脂质纳米颗粒组合物用于诱导由蛋白质表达产生的药理学作用,所述药理学作用例如,通过递送适合的红细胞生成素mrna产生增加的红细胞,或者通过递送用于编码合适的抗原或抗体的mrna来防止感染。
[0026]
本发明的脂质纳米颗粒和组合物可以用于各种目的,包括在体外和体内将封装的或缔合的(例如,复合的)诸如核酸的治疗剂递送至细胞。因此,本发明的实施方案提供通过使有此需要的对象接触封装适合的治疗剂或与适合的治疗剂缔合的脂质纳米颗粒,来治疗或预防所述对象的疾病和病症的方法,其中所述脂质纳米颗粒包含一种或多种本文描述的新型阳离子脂质。
[0027]
如本文所述,本发明的脂质纳米颗粒的实施方案对于核酸的递送是特别有用的,所述核酸包括例如mrna、反义寡核苷酸、质粒dna、微小rna(mirna)、mirna抑制剂(antagomirs/antimirs)、信使
‑
rna
‑
干扰互补rna(micrna)、dna、多价rna、dicer底物rna、互补dna(cdna)等。因此,本发明的脂质纳米颗粒和组合物可以用于通过使细胞与包含一种或多种本文描述的新型阳离子脂质的脂质纳米颗粒接触,从而在体外和体内诱导所期望蛋白质的表达,其中所述脂质纳米颗粒封装或缔合如下核酸:被表达以产生所期望蛋白质的核酸(例如,编码所期望蛋白质的信使rna或质粒)或被表达以抑制终止mrna表达的过程的核酸(例如,mirna抑制剂)。可选地,本发明的脂质纳米颗粒和组合物可以用于通过使细胞与包含一种或多种本文描述的新型阳离子脂质的脂质纳米颗粒接触,从而在体外和体内减少靶基因和靶蛋白质的表达,其中脂质纳米颗粒封装或缔合减少靶基因表达的核酸(例如,反义寡核苷酸或小干扰rna(sirna))。本发明的脂质纳米颗粒和组合物还可以用于单独或联合共递送不同的核酸(例如mrna和质粒dna),例如可以用于提供需要不同核酸(例如编码适合的基因修饰酶的mrna和并入宿主基因组的dna片段)共同定位的作用。
[0028]
可以根据任何可用的技术来制备与本发明联用的核酸。对于mrna,主要的制备方法为,但不限于酶促合成(也称为体外转录),该方法目前代表着产生长序列特异的mrna的最有效的方法。体外转录描述了由工程化的dna模板进行rna分子的模板导向合成的方法,该工程化的dna模板包含与编码目标基因的下游序列连接的上游噬菌体启动子序列(例如包括但不限于来自t7、t3和sp6大肠杆菌噬菌体)。可以用本领域熟知的适当的技术由很多来源来制备模板dna用于体外转录,所述适当的技术包括,但不限于质粒dna和聚合酶链式反应扩增(参见linpinsel,j.l和conn,g.l.,general protocols for preparation of plasmid dna template,以及bowman,j.c.,azizi,b.,lenz,t.k.,ray,p.,和williams,l.d.in rna in vitro transcription and rna purification by denaturing page in recombinant and in vitro rna syntheses methods v.941conn g.l.(编辑),new york,n.y.humana press,2012)。
[0029]
使用线性化dna模板,在相应的rna聚合酶以及腺苷、鸟苷、尿苷和胞苷核糖核苷三磷酸(rntp)存在下,在支持聚合酶活性同时使所得的mrna转录物潜在的降解最小化的条件下,在体外发生rna的转录。可以使用各种商购的试剂盒以及商购的试剂来进行体外转录,所述试剂盒包括,但不限于ribomax大规模rna生产系统(promega)、megascript转录试剂盒(lifetechnologies),以及所述试剂包括rna聚合酶和rntp。mrna的体外转录方法是本领域熟知的。(参见,例如losick,r.,1972,in vitro transcription,ann rev biochem v.41 409
‑
46;kamakaka,r.t.和kraus,w.l.2001.in vitro transcription.current protocols in cell biology.2:11.6:11.6.1
‑
11.6.17;beckert,b.和masquida,b.,(2010)synthesis of rna by in vitro transcription in rnain methods in molecular biology v.703(neilson,h.编辑),new york,n.y.humana press,2010;brunelle,j.l.和green,r.,2013,chapter five
‑
in vitro transcription from plasmid or pcr
‑
amplified dna,methods in enzymology v.530,101
‑
114;通过引用将以上全部并入本文)。
[0030]
然后从转录或相关反应的不期望的组分(包括未并入的rntp、蛋白质酶、盐、短rna寡核苷酸等)中纯化期望的体外转录的mrna。mrna转录物的分离技术是本领域熟知的。熟知的程序包括用在单价阳离子存在下的醇(乙醇、异丙醇)或氯化锂的酚/氯仿萃取或沉淀。可以使用的纯化程序的另外非限制性实例包括尺寸排阻色谱(lukavsky,p.j.和puglisi,j.d.,2004,large
‑
scale preparation and purification of polyacrylamide
‑
free rna oligonucleotides,rna v.10,889
‑
893)、基于硅的亲和色谱和聚丙烯酰胺凝胶电泳(bowman,j.c.,azizi,b.,lenz,t.k.,ray,p.,和williams,l.d.in rna in vitro transcription and rna purification by denaturing page in recombinant and in vitro rna syntheses methods v.941conn g.l.(编辑),new york,n.y.humana press,2012)。可以使用各种商购的试剂盒来进行纯化,所述试剂盒包括,但不限于sv总分离系统(promega)以及体外转录清洁和浓缩试剂盒(norgen biotek)。
[0031]
此外,虽然逆转录可以产生大量的mrna,但产物可以含有大量与不期望的聚合酶活性有关的异常rna杂质,所述杂质可能需要从全长的mrna制备物中去除。这些杂质包括由失败的转录起始导致的短rna以及由rna依赖的rna聚合酶活性、来自rna模板的rna引发的转录和自补3’延长生成的双链rna(dsrna)。已经证明这些具有dsrna结构的污染物可以通过与真核细胞中的各种先天免疫传感器相互作用而导致不期望的免疫刺激活性,所述先天免疫传感器的作用为识别特定核酸结构并诱导强效免疫应答。由于在先天细胞免疫应答期间蛋白质合成减少,因此可以进而显著减少mrna翻译。因此,已研发出用于去除这些dsrna污染物的另外的技术,并且其被本领域所知晓,包括但不限于可调比例的hplc纯化(参见例如kariko,k.,muramatsu,h.,ludwig,j.和weissman,d.,2011,generating the optimal mrna for therapy:hplc purification eliminates immune activation and improves translation of nucleoside
‑
modified,protein
‑
encoding mrna,nucl acid res,v.39e142;weissman,d.,pardi,n.,muramatsu,h.,和kariko,k.,hplc purification of in vitro transcribed long rna in synthetic messenger rna and cell metabolism modulation in methods in molecular biology v.969(rabinovich,p.h.编辑),2013)。已报导经hplc纯化的mrna以高得多的水平翻译,特别是在原代细胞中和体内。
[0032]
本领域中已描述了很多种用于改变体外转录的mrna的特定性质,并且改善其效用
的修饰。这些修饰包括,但不限于对mrna的5’末端和3’末端的修饰。内源性真核mrna通常含有在成熟分子的5
’‑
端上的帽结构,其在介导mrna帽结合蛋白质(cbp)的结合中发挥重要作用,其进而负责增强细胞中的mrna稳定性和mrna翻译效率。因此,用加帽的mrna转录物实现最高水平的蛋白质表达。5
’‑
帽含有5
’‑
多数核苷酸与鸟嘌呤核苷酸之间的5
’‑5’‑
三磷酸连接。缀合的鸟嘌呤核苷酸在n7位置被甲基化。另外的修饰包括在2
’‑
羟基上的最后的和倒数第二的多数5
’‑
核苷酸的甲基化。
[0033]
多个不同的帽结构可以用于生成体外转录合成的mrna的5
’‑
帽。合成的mrna的5
’‑
加帽可以用化学帽类似物与转录共同进行(即在体外转录过程中加帽)。例如,抗反向帽类似物(arca)帽含有5
’‑5’‑
三磷酸鸟嘌呤
‑
鸟嘌呤连接,其中一个鸟嘌呤含有n7甲基以及3
’‑
o
‑
甲基。然而,多达20%的转录物在该共转录过程中保持未加帽,并且合成的帽类似物与真实细胞mrna的5
’‑
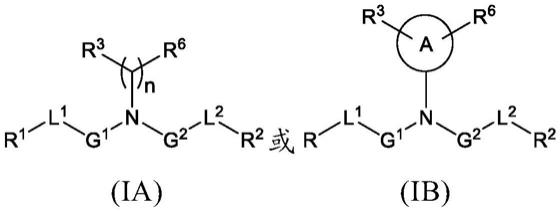
帽结构不相同,有可能降低可译性和细胞稳定性。可选地,合成的mrna分子还可以在转录后酶促加帽。这可以生成更真实的5
’‑
帽结构,其在结构上或功能上更接近地模拟内源性5
’‑
帽,该内源性5
’‑
帽具有增强的帽结合蛋白质的结合、增加的半衰期、对5’内切酶的降低的敏感性和/或减少的5’脱帽性。已研发出很多合成的5
’‑
帽类似物且被本领域所知晓,以增强mrna稳定性和可译性(参见例如,grudzien
‑
nogalska,e.,kowalska,j.,su,w.,kuhn,a.n.,slepenkov,s.v.,darynkiewicz,e.,sahin,u.,jemielity,j.,和rhoads,r.e.,synthetic mrnas with superior translation and stability properties in synthetic messenger rna and cell metabolism modulation in methods in molecular biology v.969(rabinovich,p.h.ed),2013)。
[0034]
在3
’‑
末端,通常在rna加工期间将腺嘌呤核苷酸的长链(聚腺苷酸尾)加至mrna分子。转录后,立即切割转录物的3’端以释放3’羟基,向3’端,聚腺苷酸聚合酶在被称为聚腺苷酸化的过程中将腺嘌呤核苷酸链加至rna。聚腺苷酸尾已广泛示出了增强翻译效率和mrna的稳定性(参见bernstein,p.和ross,j.,1989,poly(a),poly(a)binding protein and the regμlation of mrna stability,trends bio sci v.14373
‑
377;guhaniyogi,j.和brewer,g.,2001,regμlation of mrna stability in mammalian cells,gene,v.265,11
‑
23;dreyfus,m.and regnier,p.,2002,the poly(a)tail of mrnas:bodyguard in eukaryotes,scavenger in bacteria,cell,v.111,611
‑
613)。
[0035]
可以使用各种方法来实现体外转录的mrna的聚腺苷酸加尾,所述方法包括,但不限于,将聚(t)片段克隆至dna模板中,或者通过使用聚(a)聚合酶在转录后添加。第一种情况允许体外转录具有限定长度(取决于聚(t)片段的尺寸)的聚(a)尾的mrna,但需要对模板另外操作。后一种情况涉及使用催化腺嘌呤残基并入rna的3’末端的聚(a)聚合酶,将聚(a)尾酶促添加至体外转录的mrna,不需要对dna模板另外操作,而得到具有不同长度的聚(a)尾的mrna。可以使用各种商购的试剂盒以及商购的试剂、各种arca帽、聚(a)聚合酶等来进行5
’‑
加帽和3
’‑
聚(a)加尾,所述试剂盒包括,但不限于聚(a)聚合酶加尾试剂盒(epicenter)、mmessage mmachine t7 ultra试剂盒和聚(a)加尾试剂盒(life technologies)。
[0036]
除5’帽和3’聚腺苷酸化以外,还报导了对体外转录物的其他修饰以提供与翻译效率和稳定性有关的益处。本领域熟知的是,病原dna和rna可以通过真核细胞内的各种传感器识别,并引发强效先天免疫应答。因为天然来源的大多数核酸含有修饰的核苷,因此已显
示出辨别病原dna和rna与自身dna和rna的能力至少部分基于结构和核苷修饰。相反,体外合成的rna缺少这些修饰,因此导致其为免疫刺激的,进而可以抑制上文概述的有效的mrna翻译。将修饰的核苷引入体外转录的mrna可以用于阻止rna传感器的识别和激活,由此缓解这种不期望的免疫刺激活性,并且增强翻译能力(参见例如,kariko,k.和weissman,d.2007,naturally occurring nucleoside modifications suppress the immunostimulatory activity of rna:implication for therapeutic rna development,curr opin drug discov devel,v.10 523
‑
532;pardi,n.,muramatsu,h.,weissman,d.,kariko,k.,in vitro transcription of long rna containing modified nucleosides in synthetic messenger rna and cell metabolism modulation in methods in molecular biology v.969(rabinovich,p.h.编辑),2013);kariko,k.,muramatsu,h.,welsh,f.a.,ludwig,j.,kato,h.,akira,s.,weissman,d.,2008,incorporation of pseudouridine into mrna yields superior nonimmunogenic vector with increased translational capacity and biological stability,mol ther v.16,1833
‑
1840。用于合成修饰的rna的修饰的核苷和核苷酸可以使用本领域已知的一般方法和程序来制备、监测和使用。可使用很多种核苷修饰,其可以单独或联用其他修饰的核苷以一定程度并入体外转录的mrna(参见,例如us2012/0251618)。已报导核苷修饰的mrna的体外合成减小了激活免疫传感器的能力,伴随着增强了翻译能力。
[0037]
可修饰以在可译性和稳定性方面提供益处的mrna的其他组分包括5’和3’非翻译区(utr)。针对两者的或单独的utr优化(有利的5’和3’utr可以得自细胞或病毒rna)已显示出增强了体外转录的mrna的mrna稳定性和翻译效率(参见,例如pardi,n.,muramatsu,h.,weissman,d.,kariko,k.,in vitro transcription of long rna containing modified nucleosides in synthetic messenger rna and cell metabolism modulation in methods in molecular biology v.969(rabinovich,p.h.编著),2013)。
[0038]
除mrna以外,其他核酸净荷(payload)也可以用于本发明。对于寡核苷酸,制备方法包括但不限于化学合成和较长前体的酶促、化学裂解,上文描述的体外转录等。合成dna和rna核苷酸的方法被广泛使用,并且为本领域所熟知(参见,例如,gait,m.j.(编著)oligonucleotide synthesis:a practical approach,oxford[oxfordshire],washington,d.c.:irl press,1984;以及herdewijn,p.(编著)oligonucleotide synthesis:methods and applications,methods in molecular biology,v.288(clifton,n.j.)totowa,n.j.:humana press,2005;两者均通过引用并入本文)。
[0039]
对于质粒dna,与本发明联用的制备通常使用但不限于,在含有目标质粒的细菌的液体培养基中体外扩增和分离质粒dna。编码对特定抗生素(青霉素、卡那霉素等)的抗性的目标质粒内基因的存在允许那些含有目标质粒的细菌在含抗生素的培养基中选择性地生长。分离质粒dna的方法被广泛使用,并且为本领域所熟知(参见,例如heilig,j.,elbing,k.l.和brent,r(2001)large
‑
scale preparation of plasmid dna.current protocols in molecular biology.41:ii:1.7:1.7.1
‑
1.7.16;rozkov,a.,larsson,b.,s.,r.和schmidt,s.r.(2008),large
‑
scale production of endotoxin
‑
free plasmids for transient expression in mammalian cell culture.biotechnol.bioeng.,99:557
‑
566;以及us6197553b1)。可以使用各种可商购的试
剂盒以及可商购的试剂来进行质粒分离,所述试剂盒包括,但不限于plasmid plus(qiagen)、genjet plasmid maxiprep(thermo)和pure yield maxiprep(promega)试剂盒。
[0040]
本发明的阳离子脂质、脂质纳米颗粒和包含脂质纳米颗粒的组合物、以及它们递送诸如核酸的活性剂(例如治疗剂),以调控基因和蛋白质表达的用途的各种示例性实施方案在下文进一步详细地描述。
[0041]
如本文所使用的,除非另外指明,以下术语具有它们被赋予的含义。
[0042]
除非上下文另外需要,在本说明书和权利要求书中,词语“包含(comprise)”及其变形,如“包括”和“含有”,以开放且包括的含义解释,即,为“包括,但不限于”。
[0043]
在本说明书中,提及“一个实施方案”或“实施方案”意指结合所述实施方案描述的特定特征、结构或特性包括在本发明的至少一个实施方案中。因此,在本说明书中各处出现短语“在一个实施方案中”或“在实施方案中”不一定是全部指同一个实施方案。此外,所述特定特征、结构或特性可以以任何适合的方式与一个或多个实施方案结合。
[0044]
除非另外定义,本文使用的所有技术和科技术语具有与本发明所属领域技术人员通常理解相同的含义。如本说明书和权利要求书所使用的,除非上下文另外明确指定,单数形式“一种/一个(a)”、“一种/一个(an)”和“所述”包括复数指代物。
[0045]
短语“诱导期望蛋白质的表达”指核酸增加期望蛋白质表达的能力。为检验蛋白质表达的程度,使测试样品(例如,表达期望蛋白质的培养基中的细胞样品)或测试哺乳动物(例如,诸如人的哺乳动物或者诸如啮齿动物(例如,小鼠)或非人灵长类动物(例如,猴)模型的动物模型)接触核酸(例如,结合本发明脂质的核酸)。将测试样品或测试动物中的期望蛋白质的表达与未接触或未施用核酸的对照样品(例如,表达期望蛋白质的培养基中的细胞样品)或对照哺乳动物(例如,诸如人的哺乳动物或者诸如啮齿动物(例如,小鼠)或非人灵长类动物(例如,猴)模型的动物模型)中的期望蛋白质的表达相比较。当在对照样品或对照哺乳动物中存在期望的蛋白质时,对照样品或对照哺乳动物中期望蛋白质的表达可以指定为1.0的值。在特定的实施方案中,当测试样品或测试哺乳动物中的期望蛋白质表达与对照样品或对照哺乳动物中的期望蛋白质表达水平的比例大于1时,例如,约1.1、1.5、2.0、5.0或10.0时,实现了诱导期望蛋白质的表达。当对照样品或对照哺乳动物中不存在期望的蛋白质时,当在测试样品或测试哺乳动物中检测到任何可测水平的期望蛋白质时,实现了诱导期望蛋白质的表达。本领域的普通技术人员将理解,确定样品中蛋白质表达水平的适合的测定例如,斑点印迹、northern印迹、原位杂交、elisa、免疫沉淀、酶功能和表型测定,或者基于在适当条件下可以产生荧光或发光的报告蛋白的测定。
[0046]
短语“抑制靶基因的表达”指核酸沉默、减少或抑制靶基因表达的能力。为检验基因沉默的程度,使测试样品(例如,表达靶基因的培养基中的细胞样品)或测试哺乳动物(例如,诸如人的哺乳动物或者诸如啮齿动物(例如,小鼠)或非人灵长类动物(例如,猴)模型的动物模型)接触沉默、减少或抑制靶基因表达的核酸。将测试样品或测试动物中的靶基因的表达与未接触或未施用核酸的对照样品(例如,表达靶基因的培养基中的细胞样品)或对照哺乳动物(例如,诸如人的哺乳动物或者诸如啮齿动物(例如,小鼠)或非人灵长类动物(例如,猴)模型的动物模型)中的靶基因的表达相比较。对照样品或对照哺乳动物中的靶基因的表达可以指定为100%的值。在特定的实施方案中,当测试样品或测试哺乳动物中的靶基因表达水平相对于对照样品或对照哺乳动物中的靶基因表达水平为约95%、90%、85%、
80%、75%、70%、65%、60%、55%、50%、45%、40%、35%、30%、25%、20%、15%、10%、5%或0%时,实现了沉默、抑制或减少靶基因的表达。换言之,相对于未接触或未施用核酸的对照样品或对照哺乳动物中的靶基因表达水平,在测试样品或测试哺乳动物中,所述核酸能够沉默、减少或抑制靶基因表达的至少约5%、10%、15%、20%、25%、30%、35%、40%、45%、50%、55%、60%、65%、70%、75%、80%、85%、90%、95%或100%。确定靶基因表达水平的适合的测定包括,但不限于,使用本领域技术人员已知的技术进行的蛋白质或mrna水平的检验,所述技术例如,本领域技术人员已知的斑点印迹、northern印迹、原位杂交、elisa、免疫沉淀、酶功能以及表型测定。
[0047]
活性剂或治疗剂(例如治疗性核酸)的“有效量”或“治疗有效量”是足以产生期望效果的量,所述期望效果例如,相比不存在核酸时检测的靶序列的正常表达水平,对靶序列表达的增加或抑制。当在不存在核酸下不存在表达产物的情况下检测到任何可测量水平时,实现了靶序列表达的增加。在与核酸接触前,表达产物以一定水平存在的情况下,当使用诸如mrna的核酸得到的值的提高倍数相对于对照为约1.05、1.1、1.2、1.3、1.4、1.5、1.75、2、2.5、3、4、5、6、7、8、9、10、15、20、25、30、40、50、75、100、250、500、750、1000、5000、10000或更大时,实现了表达增加。当使用诸如反义寡核苷酸的核酸得到的值相对于对照为约95%、90%、85%、80%、75%、70%、65%、60%、55%、50%、45%、40%、35%、30%、25%、20%、15%、10%、5%或0%时,实现了靶基因或靶序列表达的抑制。测量靶基因或靶序列表达的适合的测定包括,例如,使用本领域技术人员已知的技术进行的蛋白质或rna水平的检验,所述技术例如本领域技术人员已知的斑点印迹、northern印迹、原位杂交、elisa、免疫沉淀、酶功能、适合的报告蛋白的荧光或发光以及表型测定。
[0048]
如本文所使用的,术语“核酸”指含有单链或双链形式的至少两个脱氧核糖核苷酸或核糖核苷酸的聚合物,并且包括dna、rna及其杂合体。dna可以是以下形式:反义分子、质粒dna、cdna、pcr产物或载体。rna可以是以下形式:小发夹rna(shrna)、信使rna(mrna)、反义rna、mirna、micrna、多价rna、dicer底物rna或病毒rna(vrna),及以上的组合。核酸包括含有已知核苷酸类似物或修饰的骨架残基或连接的核酸,所述核苷酸类似物或修饰的骨架残基或连接是合成的、天然存在的和非天然存在的,并且具有与参考核酸类似的结合性质。这类类似物的实例包括,但不限于,硫代磷酸酯、氨基磷酸酯、甲基磷酸酯、手性
‑
甲基磷酸酯、2
’‑
o
‑
甲基核糖核苷酸以及肽
‑
核酸(pna)。除非特别限定,该术语涵盖包含具有与参考核酸类似的结合性质的已知天然核苷酸的类似物的核酸。除非另外表明,特定核酸序列也隐含地涵盖其保守修饰的变体(例如,简并密码子取代)、等位基因、直系同源物、单核苷酸多态性、和互补序列以及明确表明的序列。特别地,可以通过生成一个或多个所选(或全部)密码子的三位被混合碱基和/或脱氧肌苷残基取代的序列来实现简并密码子取代(batzer等人,nucleic acid res.,19:5081(1991);ohtsuka等人,j.biol.chem.,260:2605
‑
2608(1985);rossolini等人,mol.cell.probes,8:91
‑
98(1994))。“核苷酸”含有糖(脱氧核糖(dna)或核糖(rna))、碱基和磷酸基团。核苷酸通过磷酸基团连接在一起。“碱基”包括嘌呤和嘧啶,其进一步包括天然化合物腺嘌呤、胸腺嘧啶、鸟嘌呤、胞嘧啶、尿嘧啶、肌苷和天然类似物,以及嘌呤和嘧啶的合成衍生物,其包括但不限于,配置新反应基团的修饰,所述新反应基团例如,但不限于,胺、醇、硫醇、羧化物和烃基卤化物。
[0049]
术语“基因”指包含产生多肽或前体多肽所必须的部分长度或全长的编码序列的
核酸(例如,dna或rna)序列。
[0050]
如本文所使用的,“基因产物”指诸如rna转录物或多肽的基因产物。
[0051]
术语“脂质”指一组有机化合物,其包括但不限于脂肪酸的酯,并且特征通常为在水中具有差的溶解性,但在很多种有机溶剂中可溶。它们通常分为至少三类:(1)“简单脂质”,其包括脂肪和油以及蜡;(2)“化合物脂质”,其包括磷脂和糖脂;以及(3)“衍生脂质”,例如类固醇。
[0052]“类固醇”为包含以下碳骨架的化合物:
[0053][0054]
类固醇的非限定性实例包括胆固醇等。
[0055]“阳离子脂质”指能够带正电的脂质。示例性的阳离子脂质包括一种或多种带有正电荷的胺基团。优选的阳离子脂质是可电离的,以便它们可以根据ph值以带正电的形式或中性形式存在。阳离子脂质的电离影响脂质纳米颗粒在不同ph条件下的表面电荷。这种电荷状态可以影响血浆蛋白质吸收、血液清除和组织分布(semple,s.c.等人,adv.drug deliv rev 32:3
‑
17(1998))以及形成核内体溶解(endosomolytic)非双层结构的能力(hafez,i.m.等人,gene ther 8:1188
‑
1196(2001)),对于核酸的细胞内递送是至关重要的。
[0056]
术语“聚合物缀合的脂质”指包含脂质部分和聚合物部分的分子。聚合物缀合的脂质的实例是聚乙二醇化脂质。术语“聚乙二醇化脂质”指包含脂质部分和聚乙二醇部分的分子。聚乙二醇化脂质为本领域已知的,并且包括1
‑
(单甲氧基
‑
聚乙二醇)
‑
2,3
‑
二肉豆蔻酰甘油(peg
‑
dmg)等。
[0057]
术语“中性脂质”指在选定的ph下以无电荷或中性两性离子形式存在的许多脂质物质中的任一种。在生理ph下,此类脂质包括但不限于磷脂酰胆碱,例如1,2
‑
二硬脂酰基
‑
sn
‑
甘油基
‑3‑
磷酸胆碱(dspc)、1,2
‑
二棕榈酰基
‑
sn
‑
甘油基
‑3‑
磷酸胆碱(dppc)、1,2
‑
二肉豆蔻酰基
‑
sn
‑
甘油基
‑3‑
磷酸胆碱(dmpc)、1
‑
棕榈酰基
‑2‑
油酰基
‑
sn
‑
甘油基
‑3‑
磷酸胆碱(popc)、1,2
‑
二油酰基
‑
sn
‑
甘油基
‑3‑
磷酸胆碱(dopc),磷脂酰乙醇胺如1,2
‑
二油酰基
‑
sn
‑
甘油基
‑3‑
磷酸乙醇胺(dope),鞘磷脂(sm),神经酰胺,类固醇如甾醇及其衍生物。中性脂质可以是合成的或天然来源的。
[0058]
术语“带电脂质”指以下多种脂质物质中的任一种,其以带正电荷或带负电荷的形式存在,不依赖于在有用的生理学范围内的ph,例如ph~3至ph~9。带电脂质可以是合成的或天然来源的。带电脂质的实例包括磷脂酰丝氨酸、磷脂酸、磷脂酰甘油、磷脂酰肌醇、甾醇半琥珀酸酯、二烷基三甲铵
‑
丙烷(例如dotap、dotma)、二烷基二甲基氨基丙烷、乙基磷酸胆碱、二甲基氨基乙烷氨基甲酰基甾醇(例如dc
‑
chol)。
[0059]
术语“脂质纳米颗粒”指具有纳米量级上(例如,1nm至1,000nm)的至少一个维度的颗粒,其包含一种或多种结构(i)的化合物或其他特定阳离子脂质。在一些实施方案中,脂质纳米颗粒包含在可用于将诸如核酸(例如,mrna)的活性剂或治疗剂递送至目标靶位点
(例如,细胞、组织、器官、肿瘤等)的制剂中。在一些实施方案中,本发明的脂质纳米颗粒包含核酸。这类脂质纳米颗粒通常包含结构(i)的化合物以及一种或多种选自中性脂质、带电脂质、胆固醇和聚合物缀合的脂质的赋形剂。在一些实施方案中,诸如核酸的活性剂或治疗剂可以封装于脂质纳米颗粒的脂质部分中,或者封装于由脂质纳米颗粒的一些或全部脂质部分包裹的水性空间中,从而保护其不被酶促降解,或者不受到由宿主生物体或细胞的机制诱导的其他不期望的作用,例如不良的免疫应答。
[0060]
在各种实施方案中,脂质纳米颗粒具有以下的平均直径:约30nm至约150nm、约40nm至约150nm、约50nm至约150nm、约60nm至约130nm、约70nm至约110nm、约70nm至约100nm、约80nm至约100nm、约90nm至约100nm、约70至约90nm、约80nm至约90nm、约70nm至约80nm、或者约30nm、35nm、40nm、45nm、50nm、55nm、60nm、65nm、70nm、75nm、80nm、85nm、90nm、95nm、100nm、105nm、110nm、115nm、120nm、125nm、130nm、135nm、140nm、145nm或150nm,并且是基本无毒的。在某些实施方案中,核酸当存在于脂质纳米颗粒时,其在水溶液中抵抗被核酸酶降解。在例如,美国专利公开第2004/0142025号、第2007/0042031号和pct公开第wo 2013/016058号和第wo 2013/086373号中公开了包含核酸的脂质纳米颗粒及其制备方法,其全部公开内容为了所有目的通过引用整体并入本文。
[0061]
如本文所使用的,“脂质封装的”指为诸如核酸(例如,mrna)的活性剂或治疗剂提供完全封装、部分封装或二者的脂质纳米颗粒。在一个实施方案中,核酸(例如,mrna)完全封装于脂质纳米颗粒中。
[0062]
如本文所使用的,术语“水溶液”指包含水的组合物。
[0063]
就核酸
‑
脂质纳米颗粒而言,“血清稳定的”意指核苷酸在暴露于血清后或在暴露于会显著降解游离dna或rna的核酸酶测定后,不会显著降解。适合的测定包括,例如,标准的血清测定、dna酶测定或rna酶测定。
[0064]
如本文所使用的,“全身性递送”指可以导致活性剂在生物体内广泛暴露的治疗性产物的递送。一些施用技术可以导致某些试剂的全身性递送,但不会全身性递送其他试剂。全身性递送意味着有用量的,优选治疗量的试剂暴露于身体的多数部位。脂质纳米颗粒的全身性递送可以通过本领域已知的任何方式进行,包括,例如,静脉内、动脉内、皮下和腹膜内递送。在一些实施方案中,脂质纳米颗粒的全身性递送通过静脉内递送。
[0065]
如本文所使用的,“局部递送”指将活性剂直接递送至生物体内的靶位点。例如,可以通过直接注入诸如肿瘤的疾病位点,诸如炎症位点的其他靶位点,或诸如肝、心脏、胰腺、肾等靶器官中来局部递送试剂。局部递送也可以包括局部应用或局部注射技术,例如肌内、皮下或皮内注射。局部递送不妨碍全身性的药理学作用。
[0066]“烃基”指仅由碳原子和氢原子组成的直链或支链的烃链基团,其为饱和的或不饱和的(即,含有一个或多个双键(烯基)和/或叁键(炔基)),具有例如,一至二十四个碳原子(c1‑
c
24
烃基)、四至二十个碳原子(c4‑
c
20
烃基)、六至十六个碳原子(c6‑
c
16
烃基)、六至九个碳原子(c6‑
c9烃基)、一至十五个碳原子(c1‑
c
15
烃基)、一至十二个碳原子(c1‑
c
12
烃基)、一至八个碳原子(c1‑
c8烃基)或一至六个碳原子(c1‑
c6烃基),并且其通过单键与分子的剩余部分连接,例如,甲基、乙基、正丙基、1
‑
甲基乙基(异丙基)、正丁基、正戊基、1,1
‑
二甲基乙基(叔丁基)、3
‑
甲基己基、2
‑
甲基己基、乙烯基、丙
‑1‑
烯基、丁
‑1‑
烯基、戊
‑1‑
烯基、戊
‑
1,4
‑
二烯基、乙炔基、丙炔基、丁炔基、戊炔基、己炔基等。除非在本说明书中另外明确说明,烃基是
任选取代的。
[0067]“亚烃基”或“亚烃基链”指将分子的剩余部分连接至自由基基团的直链或支链的二价烃链,其仅由碳和氢组成,其为饱和的或不饱和的(即,含有一个或多个双键(亚烯基)和/或叁键(亚炔基)),并且具有例如一至二十四个碳原子(c1‑
c
24
亚烃基)、一至十五个碳原子(c1‑
c
15
亚烃基)、一至十二个碳原子(c1‑
c
12
亚烃基)、一至八个碳原子(c1‑
c8亚烃基)、一至六个碳原子(c1‑
c6亚烃基)、二至四个碳原子(c2‑
c4亚烃基)、一至二个碳原子(c1‑
c2亚烃基),例如,亚甲基、亚乙基、亚丙基、亚正丁基、亚乙烯基、亚丙烯基、亚正丁烯基、亚丙炔基、亚正丁炔基等。亚烃基链通过单键或双键连接至分子的剩余部分,以及通过单键或双键连接至自由基基团。亚烃基链与分子的剩余部分和与自由基基团的连接点可以是通过链中的一个碳或任何两个碳。除非在本说明书中另外明确说明,亚烃基链可以被任选取代。
[0068]“环烃基”或“碳环”指仅由碳和氢原子组成的稳定的非芳香的单环或多环烃基,其可以包括稠合的或桥接的环系统,具有3至15个碳原子,优选具有3至10个碳原子,其为饱和的或不饱和的并且通过单键连接至分子的剩余部分。单环基团包括,例如,环丙基、环丁基、环戊基、环己基,环庚基和环辛基。多环基团包括,例如,金刚烷基、降冰片基、十氢萘基、7,7
‑
二甲基
‑
双环[2.2.1]庚烷基等。除非说明书中另有特别说明,环烃基可以为任选取代的。
[0069]“亚环烃基”为二价环烃基。除非说明书中另有特别说明,亚环烃基可以为任选取代的。
[0070]
本文使用的术语“取代的”意指任何上述基团(例如,烃基、亚烃基、环烃基或亚环烃基),其中至少一个氢原子被连有非氢原子的键取代,该非氢原子例如,但不限于:诸如f、cl、br或i的卤素原子;氧代基团(=o);羟基(
‑
oh);c1‑
c
12
烃基基团;环烃基基团;
‑
(c=o)or’;
–
o(c=o)r’;
‑
c(=o)r’;
‑
or’;
‑
s(o)
x
r’;
‑
s
‑
sr’;
‑
c(=o)sr’;
‑
sc(=o)r’;
‑
nr’r’;
‑
nr’c(=o)r’;
‑
c(=o)nr’r’;
‑
nr’c(=o)nr’r’;
‑
oc(=o)nr’r’;
‑
nr’c(=o)or’;
‑
nr’s(o)
x
nr’r’;
‑
nr’s(o)
x
r’;和
‑
s(o)
x
nr’r’,其中r’在每次出现时独立地为h、c1‑
c
15
烃基或环烃基,并且x为0、1或2。一些实施方案中,所述取代基为c1‑
c
12
烃基。在其他实施方案中,所述取代基为环烃基。在其他实施方案中,所述取代基为卤代基团,例如氟代。在其他实施方案中,所述取代基为氧代基团。在其他实施方案中,所述取代基为羟基。在其他实施方案中,所述取代基为烃氧基(
‑
or’)。在其他实施方案中,所述取代基为羧基。在其他实施方案中,所述取代基为胺基团(
‑
nr’r’)。
[0071]“任选的”或“任选地”(例如,任选取代的)意指其后描述的情况事件可发生,也可不发生,并且该描述包括所述事件或情况发生的实例以及所述事件或情况不发生的实例。例如,“任选取代的烃基”意指所述烃基可以被取代,也可以不被取代,并且该描述包括取代的烃基和无取代的烃基。
[0072]“前药”意在表明在生理条件下或通过溶剂解可以转化为本发明的生物活性化合物的化合物。因此,术语“前药”指药物可接受的本发明化合物的代谢前体。当向有需要的对象施用时,前药可以是无活性的,但在体内被转化为本发明的活性化合物。前药通常在体内快速转化得到本发明的母体化合物,例如,通过在血液中水解。所述前药化合物通常提供在哺乳动物生物体内的溶解性、组织相容性或延迟释放的优点(参见,bundgard,h.,design of prodrugs(1985),7
‑
9,21
‑
24页(elsevier,amsterdam)。在higuchi,t.等人,a.c.s.symposium series,第14卷,以及bioreversible carriers in drug design,编著
edward b.roche,american pharmaceutical association和pergamon press,1987中提供了对前药的论述。
[0073]
术语“前药”还意在包括任何共价键合的载体,当这类前药对哺乳动物对象施用时,所述载体在体内释放本发明的活性化合物。本发明化合物的前药可以通过修饰本发明化合物中存在的官能团来制备,通过这种方式,所述修饰通过常规操作或在体内裂解成为本发明的母体化合物。前药包括以下本发明化合物:其中羟基、氨基或巯基与以下的任何基团键合,当本发明化合物的前药对哺乳动物对象施用时,所述基团裂解分别形成游离羟基、游离氨基或游离巯基。前药的实例包括,但不限于,本发明化合物中胺官能团的醇或酰胺衍生物的乙酸酯、甲酸酯和苯甲酸酯衍生物等。
[0074]
本文公开的本发明还意在涵盖所有通过将一个或多个原子取代成为具有不同原子量或质量数的原子而被同位素标记的结构(i)化合物的药物可接受的化合物。可并入所公开化合物的同位素的实例包括氢、碳、氮、氧、磷、氟、氯和碘的同位素,分别例如2h、3h、
11
c、
13
c、
14
c、
13
n、
15
n、
15
o、
17
o、
18
o、
31
p、
32
p、
35
s、
18
f、
36
cl、
123
i和
125
i。这些放射性标记的化合物可以有用于:通过表征,例如,作用的位点或方式,或与药理学重要的作用位点的结合亲和力来帮助确定或测量化合物的效果。某些同位素标记的结构(i)或(ii)的化合物,例如,那些并入放射性同位素的结构(i)或(ii)的化合物,在药物和/或底物组织分布研究中是有用的。放射性同位素氚(即,3h)和碳
‑
14(即,
14
c),由于它们易于并入并且有现有的检测手段,对于这一目的是特别有用的。
[0075]
用诸如氘(即,2h)的更重的同位素取代,可以提供归因于更强的代谢稳定性的某些治疗优点,例如,增长的体内半衰期或减少的剂量需要,并且因此在一些情况下可以是优选的。
[0076]
用正电子发射同位素(例如
11
c、
18
f、
15
o和
13
n)取代,在用于检验底物受体占有率的正电子发射断层扫描(pet)研究中可以是有用的。通常可以通过本领域技术人员已知的常规技术,或者通过与下文阐述的制备和实施例中所描述类似的方法,使用适当的同位素标记的试剂代替先前使用的未标记的试剂来制备同位素标记的结构(i)的化合物。
[0077]
本文公开的本发明还意在涵盖所公开化合物的体内代谢产物。这类产物可以得自,例如,所施用化合物的,主要由于酶促过程的氧化、还原、水解、酰胺化、酯化等。因此,本发明包括由以下方法所产生的化合物,所述方法包括将本发明的化合物对哺乳动物施用足以产生其代谢产物的时间。这类产物通常通过以下方式来鉴定:将放射性标记的本发明化合物以可检测的剂量对诸如大鼠、小鼠、豚鼠、猴的动物施用,或者对人施用,持续足够的时间以允许代谢发生,并将其转化产物从尿、血液或其他生物样品分离。
[0078]“稳定的化合物”和“稳定的结构”意在表明足够稳定以便在从反应混合物分离至有用纯度以及在配制成为有效治疗剂的过程中得以保留的化合物。
[0079]“哺乳动物”包括人;以及家养动物,例如实验动物和家庭宠物(例如,猫、狗、猪、牛、绵羊、山羊、马、兔),和非家养动物,例如野生动物等。
[0080]“药物可接受的载体、稀释剂或赋形剂”包括但不限于:经美国食品药品管理局批准,用于人或家养动物可接受的任何佐剂、载体、赋形剂、助流剂、甜味剂、稀释剂、防腐剂、染色/着色剂、增味剂、表面活性剂、润湿剂、分散剂、悬浮剂、稳定剂、等渗剂、溶剂或乳化剂。
[0081]“药物可接受的盐”包括酸加成盐和碱加成盐。
[0082]“药物可接受的酸加成盐”指保留游离碱的生物效果和性质,不是生物上或其他方面不期望的,并且与以下无机酸和有机酸形成的那些盐,所述无机酸例如,但不限于盐酸、氢溴酸、硫酸、硝酸、磷酸等,而所述有机酸例如,但不限于乙酸、2,2
‑
二氯乙酸、己二酸、海藻酸、抗坏血酸、天冬氨酸、苯磺酸、苯甲酸、4
‑
乙酰氨基苯甲酸、樟脑酸、樟脑
‑
10
‑
磺酸、癸酸、己酸、辛酸、碳酸、肉桂酸、柠檬酸、环拉酸、十二烷基硫酸、乙烷
‑
1,2
‑
二磺酸、乙磺酸、2
‑
羟基乙磺酸、甲酸、富马酸、半乳糖二酸、龙胆酸、葡庚糖酸、葡糖酸、葡糖醛酸、谷氨酸、戊二酸、2
‑
氧代
‑
戊二酸、甘油磷酸、乙醇酸、马尿酸、异丁酸、乳酸、乳糖酸、月桂酸、马来酸、苹果酸、丙二酸、扁桃酸、甲磺酸、粘酸、萘
‑
1,5
‑
二磺酸、萘
‑2‑
磺酸、1
‑
羟基
‑2‑
萘甲酸、烟酸、油酸、乳清酸、草酸、棕榈酸、双羟萘酸、丙酸、焦谷氨酸、丙酮酸、水杨酸、4
‑
氨基水杨酸、癸二酸、硬脂酸、琥珀酸、酒石酸、硫氰酸、对甲苯磺酸、三氟乙酸、十一碳烯酸等。
[0083]“药物可接受的碱加成盐”指保留游离酸的生物效果和性质,不是生物上或其他方面不期望的那些盐。这些盐通过无机碱或有机碱与游离酸的加成来制备。来源于无机碱的盐包括,但不限于,钠盐、钾盐、锂盐、铵盐、钙盐、镁盐、铁盐、锌盐、铜盐、锰盐、铝盐等。优选的无机盐为铵盐、钠盐、钾盐、钙盐和镁盐。来源于有机碱的盐包括,但不限于,下列伯胺、仲胺和叔胺,取代的胺(包括天然存在的取代的胺)、环状胺和碱性离子交换树脂的盐:例如氨、异丙胺、三甲胺、二乙胺、三乙胺、三丙胺、二乙醇胺、乙醇胺、丹醇、2
‑
二甲基氨基乙醇、2
‑
二乙基氨基乙醇、二环己基胺、赖氨酸、精氨酸、组氨酸、咖啡因、普鲁卡因、海巴明(hydrabamine)、胆碱、甜菜碱、苯乙苄胺(benethamine)、苄星青霉素(benzathine)、乙二胺、葡糖胺、甲基葡糖胺、可可碱、三乙醇胺、氨丁三醇、嘌呤、哌嗪、哌啶、n
‑
乙基哌啶、聚胺树脂等。特别优选的有机碱为异丙胺、二乙胺、乙醇胺、三甲胺、二环己基胺、胆碱和咖啡因。
[0084]
结晶通常产生本发明化合物的溶剂化物。如本文所使用的,术语“溶剂化物”指包含一个或多个本发明化合物分子与一个或多个溶剂分子的聚集物。所述溶剂可以是水,在这种情况下所述溶剂化物可以是水合物。或者,所述溶剂可以是有机溶剂。因此,本发明的化合物可以以水合物存在,包括一水合物、二水合物、半水合物、倍半水合物、三水合物、四水合物等,以及以相应的溶剂化形式存在。本发明的化合物可以是真正的溶剂化物,而在其他情况下,本发明的化合物可以仅保留偶生的(adventitious)水,或者是水加上偶生溶剂的混合物。
[0085]“药物组合物”指本发明化合物与本领域通常公认用于向哺乳动物(例如,人)递送生物活性化合物的介质的制剂。因此这类介质包括所有药物可接受的载体、稀释剂或赋形剂。
[0086]“有效量”或“治疗有效量”指当对哺乳动物(优选为人)施用时,足以在哺乳动物(优选为人)中实现治疗的本发明化合物的量。构成“治疗有效量”的本发明脂质纳米颗粒的量将取决于化合物、病况及其严重性、施用方式以及待治疗哺乳动物的年龄,但可以由本领域的普通技术人员依据其自身的知识和本公开内容来常规确定。
[0087]
本文使用的“治疗(treating)”或“治疗(treatment)”涵盖针对患有相关疾病或病况的哺乳动物(优选为人)的所述相关疾病或病况的治疗,并且包括:
[0088]
(i)预防疾病或病况在哺乳动物中出现,尤其是,当这些哺乳动物倾向患有所述病况,但尚未被诊断为患有所述病况时;
[0089]
(ii)抑制所述疾病或病况,即,阻止其发展;
[0090]
(iii)减轻所述疾病或病况,即,使所述疾病或病况消退;或者
[0091]
(iv)减轻由所述疾病或病况导致的症状,即,在不解决根本疾病或病况的情况下减轻疼痛。如本文所使用的,术语“疾病”和“病况”可以交换使用,或者可以是不同的,因为特定的疾病或病况可能没有已知的病原体(因此尚未找出病因),因此尚未被认为是疾病,而仅作为不期望的病况或综合征,其中临床医生已经鉴定了或多或少特定组的症状。
[0092]
本发明的化合物或其药物可接受的盐可以含有一个或多个不对称中心,并且因此可以产生对映异构体、非对映异构体和其他立体异构形式,对于氨基酸,其可以根据绝对立体化学被定义为(r)
‑
或(s)
‑
,或者定义为(d)
‑
或(l)
‑
。本发明意在包括所有这些可能的异构体,以及其外消旋形式和光学纯的形式。光学活性的(+)和(
‑
)、(r)
‑
和(s)
‑
、或(d)
‑
和(l)
‑
异构体可以使用手性合成子或手性试剂来制备,或者使用常规技术,例如色谱法和分级结晶来拆分。用于制备/分离单一对映异构体的常规技术包括由适合的光学纯前体手性合成,或者使用,例如,手性高压液相层析法(hplc)的外消旋体(或盐或衍生物的外消旋体)的拆分。当本文描述的化合物含有烯族双键或其他几何不对称中心时,除非另外指定,意指该化合物包括e型和z型几何异构体。同样地,也意在包括所有的互变异构形式。
[0093]“立体异构体”指由相同的键键合的相同原子组成,但具有不同三维结构的化合物,其不可互变。本发明涵盖各种立体异构体及其混合物,并且包括“对映异构体”,所述对映异构体指分子互为不可重叠的(nonsuperimposeable)镜像的两个立体异构体。
[0094]“互变异构体”指质子从分子的一个原子转移至同一分子的另一个原子。本发明包括任何所述化合物的互变异构体。
[0095]
化合物
[0096]
一方面,本发明提供新型脂质化合物,其能够与其他脂质组分(例如中性脂质、带电脂质、类固醇和/或聚合物缀合的脂质)结合形成具有寡核苷酸的脂质纳米颗粒。不希望受理论束缚,认为这些脂质纳米颗粒保护寡核苷酸不在血清中降解,并且在体外和体内提供对细胞的寡核苷酸有效递送。
[0097]
在一个实施方案中,化合物具有以下结构(i):
[0098][0099]
或其药物可接受的盐、互变异构体、前药或立体异构体,其中:
[0100]
l1或l2中的一个为
‑
o(c=o)
‑
、
‑
(c=o)o
‑
、
‑
c(=o)
‑
、
‑
o
‑
、
‑
s(o)
x
‑
、
‑
s
‑
s
‑
、
‑
c(=o)s
‑
、sc(=o)
‑
、
‑
nr
a
c(=o)
‑
、
‑
c(=o)nr
a
‑
、nr
a
c(=o)nr
a
‑
、
‑
oc(=o)nr
a
‑
或
‑
nr
a
c(=o)o
‑
,并且l1或l2中的另一个为
‑
o(c=o)
‑
、
‑
(c=o)o
‑
、
‑
c(=o)
‑
、
‑
o
‑
、
‑
s(o)
x
‑
、
‑
s
‑
s
‑
、
‑
c(=o)s
‑
、sc(=o)
‑
、
‑
nr
a
c(=o)
‑
、
‑
c(=o)nr
a
‑
、nr
a
c(=o)nr
a
‑
、
‑
oc(=o)nr
a
‑
或
[0101]
‑
nr
a
c(=o)o
‑
或键;
[0102]
g1和g2各自独立地为未取代的c1‑
c
12
亚烷基或c1‑
c
12
亚烯基;
[0103]
g3为c1‑
c
24
亚烷基、c1‑
c
24
亚烯基、c3‑
c8亚环烷基、c3‑
c8亚环烯基;
[0104]
r
a
为h或c1‑
c
12
烃基;
[0105]
r1和r2各自独立地为c6‑
c
24
烷基或c6‑
c
24
烯基;
[0106]
r3为h、or5、cn、
‑
c(=o)or4、
‑
oc(=o)r4或
–
nr5c(=o)r4;
[0107]
r4为c1‑
c
12
烃基;
[0108]
r5为h或c1‑
c6烃基;并且
[0109]
x为0、1或2。
[0110]
在一些前述实施方案中,所述化合物具有以下结构(ia)或(ib)之一:
[0111][0112]
其中:
[0113]
a为3至8元环烃基或亚环烃基环;
[0114]
r6在每次出现时独立地为h、oh或c1‑
c
24
烃基;
[0115]
n为1至15的整数。
[0116]
在一些前述实施方案中,所述化合物具有结构(ia),在其他实施方案中,所述化合物具有结构(ib)。
[0117]
在前述的其他实施方案中,所述化合物具有以下结构(ic)或(id)之一:
[0118][0119]
其中y和z各自独立地为1至12的整数。
[0120]
在任何前述实施方案中,l1或l2之一为
‑
o(c=o)
‑
。例如,在一些实施方案中,l1和l2中的每一个为
‑
o(c=o)
‑
。在任何前述的一些不同的实施方案中,l1和l2各自独立地为
‑
(c=o)o
‑
或
‑
o(c=o)
‑
。例如,在一些实施方案中,l1和l2中的每一个为
‑
(c=o)o
‑
。
[0121]
在前述的一些不同的实施方案中,所述化合物具有以下结构(ie)或(if)之一:
[0122][0123]
在一些前述实施方案中,所述化合物具有以下结构(ig)、(ih)、(ii)或(ij)之一:
[0124][0125]
在一些前述实施方案中,n为2至12,例如2至8或2至4的整数。例如,在一些实施方案中,n为3、4、5或6。在一些实施方案中,n为3。在一些实施方案中,n为4。在一些实施方案中,n为5。在一些实施方案中,n为6。
[0126]
在一些其他前述实施方案中,y和z各自独立地为2至10的整数。例如,在一些实施方案中,y和z各自独立地为4至9或4至6的整数。
[0127]
在一些前述实施方案中,r6为h。在其他前述实施方案中,r6为c1‑
c
24
烃基。在其他实施方案中,r6为oh。
[0128]
在一些实施方案中,g3为未取代的。在其他实施方案中,g3为取代的。在各个不同的实施方案中,g3为直链的c1‑
c
24
亚烷基或直链的c1‑
c
24
亚烯基。
[0129]
在一些其他前述实施方案中,r1或r2或两者为c6‑
c
24
烯基。例如,在一些实施方案中,r1和r2各自独立地具有以下结构:
[0130][0131]
其中:
[0132]
r
7a
和r
7b
在每次出现时独立地为h或c1‑
c
12
烃基;并且
[0133]
a为2至12的整数,
[0134]
其中r
7a
、r
7b
和a各自被选择为使得r1和r2各自独立地包含6至20个碳原子。例如,在一些实施方案中a为5至9或8至12的整数。
[0135]
在一些前述实施方案中,至少一次出现的r
7a
为h。例如,在一些实施方案中,r
7a
在每次出现时为h。在前述的其他不同实施方案中,至少一次出现的r
7b
为c1‑
c8烃基。例如,在一些实施方案中,c1‑
c8烃基为甲基、乙基、正丙基、异丙基、正丁基、异丁基、叔丁基、正己基或正辛基。
[0136]
在不同的实施方案中,r1或r2或两者具有以下结构之一:
[0137][0138]
在一些前述实施方案中,r3为oh、cn、
‑
c(=o)or4、
‑
oc(=o)r4或
–
nhc(=o)r4。在一些实施方案中,r4为甲基或乙基。
[0139]
在各个不同的实施方案中,所述化合物具有以下表1中所示的结构之一。
[0140]
表1
[0141]
代表性化合物
[0142]
[0143]
[0144]
[0145]
[0146]
[0147]
[0148][0149]
应理解,如上文所述的结构(i)的化合物的任何实施方案,以及如上文所述的结构(i)的化合物中的任何特定取代基和/或变量,可以独立地与结构(i)的化合物的其他实施方案和/或取代基和/或变量结合,以形成上文未明确阐述的本发明的实施方案。另外,在对
于特定实施方案和/或权利要求中的任何特定r基团、l基团、g基团、a基团或变量a、n、x、y或z列出一系列取代基和/或变量的情况下,应理解,各个单独的取代基和/或变量可以从所述特定实施方案和/或权利要求中删除,并且剩余的取代基和/或变量的列举将会被认为在本发明的范围内。
[0150]
应理解,在本说明书中,所述式的取代基和/或变量的组合仅在这些贡献得到稳定化合物时才是允许的。
[0151]
在一些实施方案中,提供了包含结构(i)的化合物中的任一种或多种和治疗剂的组合物。例如,在一些实施方案中,所述组合物包含结构(i)的化合物中的任一种和治疗剂以及一种或多种选自中性脂质、类固醇和聚合物缀合的脂质的赋形剂。其他药物可接受的赋形剂和/或载体也包括在组合物的各种实施方案内。
[0152]
在一些实施方案中,中性脂质选自dspc、dppc、dmpc、dopc、popc、dope和sm。在一些实施方案中,中性脂质为dspc。在各种实施方案中,化合物与中性脂质的摩尔比为约2:1至约8:1。
[0153]
在各种实施方案中,组合物还包含类固醇或类固醇类似物。在某些实施方案中,所述类固醇或类固醇类似物为胆固醇。在这些实施方案的一些中,化合物与胆固醇的摩尔比为约5:1至1:1。
[0154]
在各种实施方案中,聚合物缀合的脂质为聚乙二醇化脂质。例如,一些实施方案包括聚乙二醇化二酰基甘油(peg
‑
dag)例如1
‑
(单甲氧基
‑
聚乙二醇)
‑
2,3
‑
二肉豆蔻酰基甘油(peg
‑
dmg)、聚乙二醇化磷脂酰乙醇胺(peg
‑
pe)、peg琥珀酸二酰基甘油(peg
‑
s
‑
dag)例如4
‑
o
‑
(2’,3
’‑
二(十四酰基氧基)丙基
‑1‑
o
‑
(ω
‑
甲氧基(聚乙氧基)乙基)丁二酸酯(盐)(peg
‑
s
‑
dmg)、聚乙二醇化神经酰胺(peg
‑
cer)、或peg二烷氧基丙基氨基甲酸酯例如ω
‑
甲氧基(聚乙氧基)乙基
‑
n
‑
(2,3
‑
二(十四烷氧基)丙基)氨基甲酸酯或2,3
‑
二(十四烷氧基)丙基
‑
n
‑
(ω
‑
甲氧基(聚乙氧基)乙基)氨基甲酸酯。在各种实施方案中,化合物与聚乙二醇化脂质的摩尔比为约100:1至约20:1。
[0155]
在一些实施方案中,所述组合物包含具有以下结构(ii)的聚乙二醇化脂质:
[0156][0157]
或其药物可接受的盐、互变异构体或立体异构体,其中:
[0158]
r8和r9各自独立地为含有10至30个碳原子的直链或支链的、饱和或不饱和的烃基链,其中烃基链任选地被一个或多个酯键中断;并且
[0159]
w具有30至60的平均值。
[0160]
在一些实施方案中,r8和r9各自独立地为含有12至16个碳原子的直链的、饱和的烃基链。在一些实施方案中,w具有43至53的平均值。在其他实施方案中,平均值w为约45。在其他不同的实施方案中,平均值w为约49。
[0161]
在一些前述组合物的实施方案中,治疗剂包括核酸。例如,在一些实施方案中,所述核酸选自反义rna和信使rna。
[0162]
在其他不同的实施方案中,本发明涉及向有需要的患者施用治疗剂的方法,该方法包括制备或提供上述组合物中的任一种并向患者施用该组合物。
[0163]
出于施用的目的,本发明的化合物(通常是脂质纳米颗粒与治疗剂结合的形式)可以以粗化学品施用,或者可以配制为药物组合物。本发明的药物组合物包含结构(i)的化合物和一种或多种药物可接受的载体、稀释剂或赋形剂。结构(i)的化合物以有效形成脂质纳米颗粒并递送治疗剂,例如,用于治疗相关的特定疾病或病况的量存在于组合物中。本领域技术人员可以容易地确定适当的浓度和剂量。
[0164]
本发明组合物的施用可以通过任何用于类似效用的试剂的可接受施用方式来进行。本发明的药物组合物可以配制成固体、半固体、液体或气体形式的制剂,例如片剂、胶囊、粉末、颗粒、软膏、溶液、悬浮液、栓剂、注射剂、吸入剂、凝胶、微球和气溶胶。施用这类药物组合物的典型途径包括,但不限于,口服、局部、经皮、吸入、胃肠外、舌下、口含、直肠、阴道和鼻内途径。本文使用的术语胃肠外包括皮下注射,静脉内、肌内、皮内、胸骨内注射或输注技术。配制本发明的药物组合物以便允许经过对患者施用该组合物后其中含有的活性成分是生物可利用的。待向对象或患者施用的组合物是一个或多个剂量单位的形式,其中,例如,片剂可以是单剂量单位,而本发明气溶胶形式的化合物的容器可以容纳多个剂量单位。制备这些剂型的现行的方法是已知的,或者对于本领域技术人员是显而易见的;例如,参见remington:the science and practice of pharmacy,第20版(philadelphia college of pharmacy and science,2000)。在任何情况下,待施用的组合物将会含有治疗有效量的本发明化合物或其药物可接受的盐,以便根据本发明的教导治疗相关的疾病或病况。
[0165]
本发明的药物组合物可以是固体或液体的形式。一方面,载体是微粒,使得组合物是,例如,片剂或粉末形式。载体可以是液体,此时组合物是,例如,口服糖浆、可注射液体或气溶胶,所述气溶胶适用于,例如,吸入施用。
[0166]
当意图用于口服施用时,药物组合物优选为固体或液体形式,其中本文认为是固体或液体的形式包括半固体、半液体、悬浮液和凝胶形式。
[0167]
作为用于口服施用的固体组合物,药物组合物可以配制成粉末、颗粒、压缩的片剂、丸剂、胶囊、咀嚼胶、薄片等形式。这类固体组合物通常将含有一种或多种惰性稀释剂或可食用载体。另外,可以存在以下的一种或多种:粘合剂,例如羧甲基纤维素、乙基纤维素、微晶纤维素、黄蓍胶或明胶;赋形剂,例如淀粉、乳糖或糊精;崩解剂,例如海藻酸、海藻酸钠、primogel、玉米淀粉等;润滑剂,例如硬脂酸镁或sterotex;助流剂,例如胶体二氧化硅;甜味剂,例如蔗糖或糖精;调味剂,例如薄荷、水杨酸甲酯或橙味调味剂;以及着色剂。
[0168]
当药物组合物是胶囊(例如,明胶胶囊)形式时,其可以含有上述类型材料之外的液体载体,例如聚乙二醇或油。
[0169]
药物组合物可以是液体的形式,例如,酏剂、糖浆、溶液、乳液或悬浮液。作为两种实例,液体可以用于口服施用或用于注射递送。当意图用于口服施用时,优选的组合物含有,除本发明化合物之外的,甜味剂、防腐剂、染色/着色剂和增味剂中的一种或多种。在通过注射施用的组合物中,可以包括表面活性剂、防腐剂、润湿剂、分散剂、悬浮剂、缓冲剂、稳定剂和等渗剂中的一种或多种。
[0170]
本发明的液体药物组合物,不论其为溶液、悬浮液还是其他类似的形式,可以包括以下佐剂中的一种或多种:无菌稀释剂,例如注射用水、盐水溶液、优选生理盐水、林格氏溶
液、等渗氯化钠;不挥发性油类,例如可用作溶剂或悬浮介质的合成的单甘酯或双甘酯,聚乙二醇、甘油、丙二醇或其他溶剂;抗菌剂,例如苄醇或尼泊金甲酯;抗氧化剂,例如抗坏血酸或亚硫酸氢钠;螯合剂,例如乙二胺四乙酸;缓冲剂,例如乙酸盐、柠檬酸盐或磷酸盐;以及用于调节张力的试剂,例如氯化钠或葡萄糖;用作冷冻保护剂的试剂,例如蔗糖或海藻糖。胃肠外制剂可以封装在玻璃或塑料制作的安瓿、一次性注射器或多剂量瓶中。生理盐水是优选的佐剂。可注射的药物组合物优选为无菌的。
[0171]
意图用于胃肠外施用或口服施用的本发明的液体药物组合物应含有可获得适合剂量的本发明化合物的量。
[0172]
本发明的药物组合物可以意图用于局部施用,在这种情况下,载体可以适当地包含溶液基质、乳液基质、软膏基质或凝胶基质。例如,所述基质可以包含以下的一种或多种:矿脂、羊毛脂、聚乙二醇、蜂蜡、矿物油、诸如水和醇的稀释剂、以及乳化剂和稳定剂。增稠剂可以存在于用于局部施用的药物组合物中。如果意图用于经皮施用,组合物可以包括经皮贴片或离子电渗装置。
[0173]
本发明的药物组合物可以意图用于直肠施用,例如,为栓剂的形式,其在直肠中溶解并释放药物。用于直肠施用的组合物可以含有油脂性基质作为适合的无刺激性赋形剂。这类基质包括但不限于羊毛脂、可可脂和聚乙二醇。
[0174]
本发明的药物组合物可以包括修饰固体或液体剂量单位的物理形式的各种材料。例如,组合物可以包括形成活性成分周围的包衣壳的材料。形成包衣壳的材料通常是惰性的,并且可以选自,例如,糖、虫胶、和其他肠溶包衣试剂。或者,活性成分可以封装在明胶胶囊中。
[0175]
固体或液体形式的本发明药物组合物可以包括与本发明化合物结合并由此有助于所述化合物递送的试剂。可以以这种能力作用的适合的试剂包括单克隆或多克隆抗体或蛋白质。
[0176]
本发明的药物组合物可以由可作为气溶胶施用的剂量单位组成。术语气溶胶用于表示从胶体性质的系统到由加压包装组成的系统的各种系统。可以通过液化气或压缩气来递送,或者通过分散活性成分的适合的泵系统来递送。本发明化合物的气溶胶可以以单相、双相系统或三相系统来递送,以便递送活性成分。气溶胶的递送包括必要的容器、活化剂、阀、子容器等,其在一起可以形成试剂盒。本领域技术人员不需过度实验即可确定优选的气溶胶。
[0177]
本发明的药物组合物可以通过制药领域熟知的方法来制备。例如,意图通过注射施用的药物组合物可以通过将本发明的脂质纳米颗粒与无菌、蒸馏的水或其他载体结合以便形成溶液来制备。可以加入表面活性剂以促进形成均匀的溶液或悬浮液。表面活性剂是与本发明化合物非共价地相互作用以便促进所述化合物在水性递送系统中溶解或均匀悬浮的化合物。
[0178]
本发明的组合物或其药物可接受的盐以治疗有效量施用,所述量将会根据多种因素变化,包括使用的具体治疗剂的活性;治疗剂的代谢稳定性和作用时长;患者的年龄、体重、一般健康状况、性别和饮食;施用的方式和时间;排泄速率;药物组合;具体病症或病况的严重性;以及经受治疗的对象。
[0179]
本发明的组合物也可以在施用一种或多种其他治疗剂的同时、之前或之后施用。
这类组合治疗包括施用本发明组合物和一种或多种另外的活性剂的单一药物剂量制剂,以及施用本发明组合物和各个在其自身单独药物剂量制剂中的活性剂。例如,本发明组合物和其他活性剂可以以单一口服剂量组合物(例如片剂或胶囊)一起向患者施用,或者各个试剂以不同的口服剂量制剂施用。当使用不同的剂量制剂时,本发明化合物和一种或多种另外的活性剂可以在基本同一时间(即,同时)施用,或者在相互交错的时间(即,依序)施用;应理解组合治疗包括所有的这些给药方案。
[0180]
上述化合物和组合物的制备方法在下文描述,和/或在本领域已知。
[0181]
本领域技术人员将会认识到,在本文描述的方法中,中间化合物的官能团可能需要通过适合的保护基保护。这类官能团包括羟基、氨基、巯基和羧酸。用于羟基的适合的保护基包括三烷基甲硅烷基或二芳基烷基甲硅烷基(例如,叔丁基二甲基甲硅烷基、叔丁基二苯基甲硅烷基或三甲基甲硅烷基)、四氢吡喃基、苄基等。用于氨基、脒基和胍基的适合的保护基包括叔丁氧基羰基、苄氧基羰基等。用于巯基的适合的保护基包括
‑
c(o)
‑
r”(其中r”为烃基、芳基或芳烃基)、对甲氧基苄基、三苯甲基等。用于羧酸的适合的保护基包括烃基、芳基或芳烃基酯。保护基可以根据标准技术添加或去除,所述标准技术是本领域技术人员已知的和本文中描述的。在green,t.w.和p.g.m.wutz,protective groups in organic synthesis(1999),第3版,wiley中详细描述了保护基团的使用。正如本领域技术人员会认识到的,保护基也可以是聚合物树脂,例如wang树脂、rink树脂或2
‑
氯三苯甲基
‑
氯树脂。
[0182]
本领域技术人员还将认识到,虽然本发明化合物的这类经保护的衍生物可以不由此具有药理活性,但其可以对哺乳动物施用并且之后在体内代谢形成具有药理活性的本发明化合物。这类衍生物因此可以被描述为“前药”。所有本发明化合物的前药包括在本发明的范围内。
[0183]
此外,所有以游离碱或游离酸形式存在的本发明化合物可以根据本领域技术人员已知的方法用适当的无机或有机的碱或酸处理来转化为其药物可接受的盐。本发明化合物的盐可以通过标准技术转化为其游离碱或酸形式。
[0184]
以下通用反应方案1示例性说明了制备本发明化合物的方法,所述本发明化合物即结构(i)的化合物
[0185][0186]
或其药物可接受的盐、互变异构体或立体异构体,其中r1、r2、r3、l1、l2、g1、g2和g3如本文所定义。应理解,本领域技术人员能够通过类似的方法或者通过结合本领域技术人员已知的其他方法来制备这些化合物。还应理解,本领域技术人员能够以下文描述的类似的方式,通过使用适当的起始组分并按照需要修改合成的参数,来制备下文未明确说明的结构(i)的其他化合物。通常,起始组分可以得自诸如sigma aldrich、lancaster synthesis,inc.、maybridge、matrix scientific、tci和fluorochem usa等来源,或者根据本领域技术人员已知的来源合成(参见,例如,advanced organic chemistry:reactions,mechanisms,and structure,第5版(wiley,2000年12月)),或者如本发明描述来制备。
[0187]
通用反应方案1
[0188][0189]
通用反应方案i提供了用于制备结构(i)的化合物的示例性方法。通用反应方案中的g1、g3、r1和r3如本文所定义,并且g1’指g1的一个碳的较短的同系物。购买结构a
‑
1的化合物,或根据本领域已知的方法制备结构a
‑
1的化合物。在适当的缩合条件下(例如,dcc)a
‑
1与二醇a
‑
2的反应产生了酯/醇a
‑
3,然后可以将其氧化(例如,pcc)成醛a
‑
4。在还原胺化条件下a
‑
4与胺a
‑
4的反应产生结构(i)的化合物。
[0190]
应注意到对于本领域普通技术人员而言可获得用于制备结构(i)的化合物的各种替代策略。例如,可以根据类似的方法使用适当的原材料制备其中l1和l2不同于酯的其他的结构(i)的化合物。此外,通用反应方案1描述了结构(i)的化合物的制备,其中g1和g2相同;然而,这不是本发明的必需方面,并且对上述反应方案的修改可以产生其中g1和g2是不同的化合物。根据需要使用保护基和对上述通用反应方案的其他修改对于本领域普通技术人员而言会是显而易见的。
[0191]
提供了以下实施例,其目的在于例示而并非限定。
[0192]
实施例1
[0193]
利用脂质纳米颗粒组合物的荧光素酶mrna体内评价将阳离子脂质、dspc、胆固醇和peg
‑
脂质以50:10:38.5:1.5或47.5:10:40.8:1.7的摩尔比溶解在乙醇中。以约10:1至30:1的总脂质与mrna的重量比制备脂质纳米颗粒(lnp)。简而言之,将mrna在10mm至50mm柠檬酸盐缓冲液(ph4)中稀释至0.2mg/ml。使用注射器泵,将脂质的乙醇溶液与mrna水溶液以约1:5至1:3(体积/体积)的比例混合,总流速为15ml/min以上。然后去除乙醇,并通过透析用pbs替代外部的缓冲液。最后,将脂质纳米颗粒通过0.2μm孔的无菌过滤器过滤。使用malvern zetasizer nano zs(malvern,uk)通过准弹性光散射测定的脂质纳米颗粒的粒径为直径大约55
‑
95nm,并且在一些情况下,直径大约70
‑
90nm。
[0194]
根据实验动物管理委员会(acc)和加拿大动物管理委员会(ccac)制定的指南,在6
‑
8周龄的雌性c57bl/6小鼠(charles river),8
‑
10周龄cd
‑
1(harlan)小鼠(charles river)上进行研究。通过尾静脉注射全身性给予不同剂量的mrna
‑
脂质纳米颗粒,并在给药后的特定时间点(例如4小时)使动物安乐死。将肝脏和脾脏收集在预先称重的管中,确定重量,立即在液氮中快速冷冻,并且在
‑
80℃下储存,直至用于分析。对于肝脏,切割约50mg以便在2mlfastprep管(mp biomedicals,solon oh)中进行分析。向各个管中加入1/4”陶瓷球(mp biomedicals),并将平衡至室温的500μl的glo裂解缓冲液
‑
glb(promega,madison wi)加入到肝脏组织中。使用fastprep24仪器(mp biomedicals)将肝脏组织在2x 6.0m/s下均匀化15秒。将匀浆在室温下孵育5分钟,然后在glb中进行1:4稀释,并使用steadyglo荧光素
酶测定系统(promega)进行评估。具体地,将50μl的稀释的组织匀浆与50μl的steadyglo底物反应,摇振10秒,接着孵育5分钟,然后使用centroxs3 lb 960光度计(berthold technologies,germany)定量。通过使用bca蛋白质测定试剂盒(pierce,rockford il)来确定测定的蛋白质的量。然后将相对发光度单位(rlu)归一化成所测定蛋白质的总μg。为了将rlu转化成ng荧光素酶,用quantilum重组荧光素酶(promega)生成了标准曲线。基于图1中提供的数据,选择了四小时时间点用于脂质制剂的效能评价。
[0195]
来自trilink biotechnologies的fluc mrna(l
‑
6107)将表达荧光素酶蛋白,其最初从萤火虫(photinus pyralis)中分离出来。fluc通常用于哺乳动物细胞培养物中以测量基因表达和细胞活力。其在底物萤光素存在下发射出生物性光。这种加帽并且聚腺苷酸化的mrna被5
‑
甲基胞苷和假尿苷完全取代。
[0196]
实施例2
[0197]
所配制的脂质的pk
a
的测定如本文其他部分所述,所配制的阳离子脂质的pka与用于递送核酸的lnp的效果相关(参见jayaraman等人,angewandte chemie,国际版(2012),51(34),8529
‑
8533;semple等人,nature biotechnology 28,172
‑
176(2010))。优选的pka范围是~5至~7。使用基于2
‑
(对甲苯胺基)
‑6‑
萘磺酸(tns)的荧光的分析,在脂质纳米颗粒中测定各阳离子脂质的pk
a
。如实施例1中所述,使用有序的方法来制备在pbs中的浓度为0.4mm总脂质的包含阳离子脂质/dspc/胆固醇/peg
‑
脂质(50/10/38.5/1.5mol%)的脂质纳米颗粒。将tns在蒸馏水中制备成100μm储备溶液。将囊泡稀释成在2ml缓冲溶液中含24μm脂质,所述缓冲溶液含有10mm hepes、10mm mes、10mm乙酸铵、130mm nacl,其中ph为2.5至11。加入等份的tns溶液以产生1μm的终浓度,并且在涡旋混合之后,在室温下使用321nm和445nm的激发波长和发射波长在slm aminco series 2发光分光光度计中测量荧光强度。对荧光数据应用s形最佳拟合分析,并将pk
a
测量为产生半数最大荧光强度的ph(参见图2)。
[0198]
实施例3
[0199]
使用体内荧光素酶mrna表达的啮齿动物模型测定含有各种阳离子脂质的脂质纳米颗粒制剂的效能之前用核酸测试了表2中示出的阳离子脂质。为了比较的目的,如实施例1和pct/us10/22614中所述(将其据此通过引用整体并入本文),使用有序(in line)混合方法,将这些脂质也用于配制含有fluc mrna(l
‑
6107)的脂质纳米颗粒。使用以下摩尔比来配制脂质纳米颗粒:50%阳离子脂质/10%二硬脂酰磷脂酰胆碱(dspc)/38.5%胆固醇/1.5%peg脂质(“peg
‑
dmg”,即,(1
‑
(单甲氧基
‑
聚乙二醇)
‑
2,3
‑
二肉豆蔻酰基甘油,平均peg分子量为2000)。如实施例1所述,在经由尾静脉注射施用之后的4小时,通过测量肝脏中的荧光素酶表达来确定相对活性。在0.3和1.0mg mrna/kg的剂量下比较所述活性,并表达成在如实施例1所述的施用之后4小时测量的ng荧光素酶/g肝脏。
[0200]
表2
[0201]
与mrna表现出活性的比较脂质
[0202][0203][0204]
使用以下摩尔比来配制表3中示出的本发明的代表性化合物:a)50%阳离子脂质/10%二硬脂酰磷脂酰胆碱(dspc)/38.5%胆固醇/1.5%peg脂质(“peg
‑
dma”2
‑
[2
‑
(ω
‑
甲氧基(聚乙二醇
2000
)乙氧基]
‑
n,n
‑
双十四烷基乙酰胺)或b)47.5%阳离子脂质/10%dspc/40.8%胆固醇/1.7%peg脂质。如实施例1所述,在经由尾静脉注射给药之后4小时,通过测量肝脏中的荧光素酶表达来测定相对活性。在0.3和1.0mg mrna/kg的剂量下比较所述活性,并且表达成如实施例1所述在给药之后4小时测量的ng荧光素酶/g肝脏。图3中给出了所选数据的图(从上到下:三角形=化合物3;圆形=化合物2;十字形=化合物1;正方形=mc3)。
[0205]
表3
[0206]
新型阳离子脂质及相关活性
[0207]
[0208]
[0209]
[0210][0211]
实施例4
[0212]6‑
(2
’‑
己基癸酰基氧基)己
‑1‑
醛的合成
[0213]
用2
‑
己基癸酸(19.8g)、dcc(18.2g)和dmap(11.3g)处理己
‑
1,6
‑
二醇(27.6g)的二氯甲烷(475ml)溶液。将溶液搅拌三天。过滤反应混合物并向滤液中添加己烷(500ml)。搅拌混合物并使沉淀物沉淀出。倾析出上清液并用稀盐酸洗涤。将有机相经无水硫酸镁干燥、过滤并去除溶剂,产生30g粗产物。
[0214]
将粗产物溶于二氯甲烷(200ml)中,并用氯铬酸吡啶鎓盐(15g)处理两小时。添加乙醚(600ml)并通过硅胶床过滤上清液。从滤液中去除溶剂,并将所得的油溶于己烷中。通过硅胶塞过滤悬浮液并去除溶剂。使残留物通过硅胶柱(80g),使用己烷随后使用二氯甲烷作为洗脱液。获得呈无色油的6
‑
(2
’‑
己基癸酰基氧基)己
‑1‑
醛(24g)。
[0215]
实施例5
[0216]4‑
(2
’‑
己基癸酰基氧基)丁
‑1‑
醛的合成
[0217]
用2
‑
己基癸酸(9.2g)、dcc(8.8g)和dmap(4.9g)处理丁
‑
1,4
‑
二醇(12.5g)的二氯甲烷(200ml)溶液。搅拌溶液过夜。过滤反应混合物,并去除溶剂。将残留物溶于二氯甲烷中并用稀盐酸洗涤。将有机相经无水硫酸镁干燥,通过硅胶床过滤并去除溶剂。
[0218]
将粗产物溶于二氯甲烷(150ml)中,并用氯铬酸吡啶鎓盐(6g)处理一小时。添加乙醚(450ml)并通过硅胶床过滤上清液。从滤液中去除溶剂,并将所得的油溶于己烷中。通过硅胶床过滤悬浮液并去除溶剂,产生呈无色油的4
‑
(2
’‑
己基癸酰基氧基)丁
‑1‑
醛(11g)。
[0219]
实施例6
[0220]
化合物1的合成
[0221]
将6
‑
(2
’‑
己基癸酰基氧基)己
‑1‑
醛(3.0g)、乙酸(0.21g)和乙醇胺(0.14g)的二氯甲烷(50ml)溶液用三乙酰氧基硼氢化钠(1.4g)处理过夜。用稀氢氧化钠水溶液洗涤该溶液。将有机相经无水硫酸镁干燥、过滤并去除溶剂。使用甲醇/二氯甲烷(0
‑
8/100
‑
92%)梯度,使残余物通过硅胶柱,产生呈无色油的化合物1(0.63g)。
[0222]
实施例7
[0223]
化合物2的合成
[0224]
将6
‑
(2
’‑
己基癸酰基氧基)己
‑1‑
醛(3.0g)、乙酸(0.33g)和3
‑
氨基丙
‑1‑
醇(0.17g)的二氯甲烷(20ml)溶液用三乙酰氧基硼氢化钠(1.3g)处理一小时。用稀氢氧化钠水溶液洗涤该溶液。将有机相经无水硫酸镁干燥、过滤并去除溶剂。使用甲醇/二氯甲烷(0
‑
8/100
‑
92%)梯度,使残余物通过硅胶柱,产生呈无色油的化合物2(1.1g)。
[0225]
实施例8
[0226]
化合物3的合成
[0227]
将6
‑
(2
’‑
己基癸酰基氧基)己
‑1‑
醛(2.4g)、乙酸(0.33g)和4
‑
氨基丁
‑1‑
醇(0.23g)的二氯甲烷(20ml)溶液用三乙酰氧基硼氢化钠(1.3g)处理两小时。用碳酸氢钠水溶液洗涤该溶液。将有机相经无水硫酸镁干燥、过滤并去除溶剂。使用甲醇/二氯甲烷(0
‑
8/100
‑
92%)梯度,使残余物通过硅胶柱,产生呈无色油的化合物3(0.4g)。
[0228]
实施例9
[0229]
化合物4的合成
[0230]
将4
‑
(2
’‑
己基癸酰基氧基)丁
‑1‑
醛(2.4g)、乙酸(0.30g)和4
‑
氨基丁
‑1‑
醇(0.22g)的二氯甲烷(20ml)溶液用三乙酰氧基硼氢化钠(1.3g)处理两小时。用稀氢氧化钠水溶液洗涤该溶液。将有机相经无水硫酸镁干燥、过滤并去除溶剂。使用甲醇/二氯甲烷(0
‑
8/100
‑
92%)梯度,使残余物通过硅胶柱。使用乙酸/甲醇/二氯甲烷(2
‑
0/0
‑
10/98
‑
90%)梯度,使部分纯化的级分通过第二柱。用碳酸氢钠水溶液洗涤纯化的级分,产生呈无色油的化合物4(0.9g)。
[0231]
实施例10
[0232]
化合物5的合成
[0233]
将4
‑
(2
’‑
己基癸酰基氧基)丁
‑1‑
醛(2.4g)、乙酸(0.31g)和3
‑
氨基丙
‑1‑
醇(0.17g)的二氯甲烷(20ml)溶液用三乙酰氧基硼氢化钠(1.4g)处理一小时。用碳酸氢钠水溶液洗涤该溶液。将有机相经无水硫酸镁干燥、过滤并去除溶剂。使用甲醇/二氯甲烷(0
‑
8/100
‑
92%)梯度,使残余物通过硅胶柱。使用乙酸/甲醇/二氯甲烷(2
‑
0/0
‑
8/98
‑
92%)梯度,使部分纯化的级分通过第二柱。用碳酸氢钠水溶液洗涤纯化的级分,产生呈无色油的化合物5(0.57g)。
[0234]
实施例11
[0235]
化合物6的合成
[0236]
将4
‑
(2
’‑
己基癸酰基氧基)丁
‑1‑
醛(2.4g)、乙酸(0.30g)和乙醇胺(0.14g)的二氯甲烷(20ml)溶液用三乙酰氧基硼氢化钠(1.3g)处理两小时。用碳酸氢钠水溶液洗涤该溶液。将有机相经无水硫酸镁干燥、过滤并去除溶剂。使用甲醇/二氯甲烷(0
‑
10/100
‑
90%)梯度,使残余物通过硅胶柱。使用乙酸/甲醇/二氯甲烷(2
‑
0/0
‑
9/98
‑
92%)梯度,使部分纯化的级分通过第二柱。用碳酸氢钠水溶液洗涤纯化的级分,产生呈无色油的化合物6(0.2g)。
[0237]
实施例12
[0238]
化合物7的合成
[0239]
将6
‑
(2
’‑
己基癸酰基氧基)己
‑1‑
醛(2.4g)、乙酸(0.14g)和5
‑
氨基戊
‑1‑
醇(0.24g)的二氯甲烷(20ml)溶液用三乙酰氧基硼氢化钠(1.3g)处理两小时。用碳酸氢钠水溶液洗涤该溶液。将有机相经无水硫酸镁干燥、过滤并去除溶剂。使用甲醇/二氯甲烷(0
‑
8/100
‑
92%)梯度,使残余物通过硅胶柱,产生呈无色油的化合物7(0.5g)。
[0240]
实施例13
[0241]
化合物8的合成
[0242]
将6
‑
(2
’‑
己基癸酰基氧基)己
‑1‑
醛(2.4g)、乙酸(0.17g)和6
‑
氨基己
‑1‑
醇(0.26g)的二氯甲烷(20ml)溶液用三乙酰氧基硼氢化钠(1.3g)处理两小时。用碳酸氢钠水溶液洗涤该溶液。将有机相经无水硫酸镁干燥、过滤并去除溶剂。使用甲醇/二氯甲烷(0
‑
8/100
‑
92%)梯度,使残余物通过硅胶柱,产生呈无色油的化合物8(0.5g)。
[0243]
实施例14
[0244]
化合物9的合成
[0245]
将6
‑
(2
’‑
己基癸酰基氧基)己
‑1‑
醛(2.4g)和反式
‑2‑
氨基环己醇盐酸盐(0.35g)的二氯甲烷(10ml)/四氢呋喃(10ml)溶液用三乙酰氧基硼氢化钠(1.3g)处理1.5小时。用碳酸氢钠水溶液洗涤该溶液。将有机相经无水硫酸镁干燥、过滤并去除溶剂。使用甲醇/二氯甲烷(0
‑
8/100
‑
92%)梯度,使残余物通过硅胶柱,产生呈无色油的化合物9(0.6g).
[0246]
实施例15
[0247]
化合物10的合成
[0248]
向2
‑
氨基乙醇(106mg,1.75mmol)的无水thf(15ml)溶液中添加2
‑
辛基十二烷基6
‑
溴己酸酯(2当量,1.66g,3.5mmol)、碳酸钾(2当量,3.5mmol,477mg,)和碳酸铯(0.3当量,0.525mmol,171mg,),并在63c(油浴)加热16h。向混合物中添加痕量的四丁基碘化铵,并将混合物再加热回流4天。在减压下蒸发溶剂,将残余物置于己烷和乙酸乙酯(大约9:1)的混合物中并用水和盐水洗涤。将有机层分离,经无水硫酸钠干燥、过滤并在减压下蒸发,获得油(1.6g)。将残余物(1.6g)在硅胶上通过柱色谱法(meoh于氯仿中,0至4%)纯化。这产生呈无色油的化合物10(700mg,0.82mmol,47%)。
[0249]
实施例16
[0250]
化合物11的合成
[0251]
向2
‑
氨基乙醇(116mg,1.9mmol,115ul)的15ml无水thf溶液中添加2
‑
己基癸基6
‑
溴己酸酯(1.9当量,1.52g,3.62mmol)、碳酸钾(1.9当量,3.62mmol,500mg)、碳酸铯(0.3当量,0.57mmol,186mg)和碘化钠(10mg),并在ar下加热回流6天。将溶剂在减压下蒸发,将残余物置于己烷中,并用水和盐水洗涤。将有机层分离,经无水硫酸钠干燥、过滤并在减压下蒸发,获得无色油。将粗产物在硅胶上通过快速柱色谱法(meoh于氯仿中,0至4%)纯化,产
生呈无色油的化合物11(936mg,1.27mmol,70%)。
[0252]
实施例17
[0253]
化合物12的合成
[0254]
以与化合物11的程序类似的方式制备化合物12,产生538mg无色油(0.86mmol,57%)。
[0255]
实施例18
[0256]
化合物13的合成
[0257]
向2
‑
氨基乙醇(171mg,2.81mmol,169ul)的无水thf(30ml)溶液中添加2
‑
辛基十二烷基4
‑
溴丁酸酯(1.9当量,2.386g,5.33mmol)、碳酸钾(1.9当量,5.33mmol,736mg)、碳酸铯(0.3当量,0.84mmol,275mg)和碘化钠(10mg),并在ar下加热回流16h。tlc(己烷/乙酸乙酯=9:1,chcl3/meoh=19:1)显示产生显著量的2
‑
辛基
‑1‑
十二烷醇。冷却并过滤混合物。浓缩滤液,并将残余物溶于2
‑
辛基
‑1‑
十二烷醇(2.1g)中。添加一些4a分子筛珠粒和n,n
‑
二异丙基乙胺(1.9当量,5.33mmol,683mg,0.92ml)。将混合物密封,并在62c下再加热4天。冷却反应混合物。添加己烷。将己烷溶液倾析出并浓缩至干。将残余物在硅胶上通过柱色谱法(meoh于氯仿中,0至4%)纯化,产生呈无色油的化合物13(282mg,0.35mmol,13%)。
[0258]
实施例19
[0259]
化合物14的合成
[0260]
向十七烷
‑9‑
基6
‑
溴己酸酯(2当量,1.13g,2.61mmol)的无水thf(15ml)溶液中添加2
‑
氨基乙醇(1eq。1.31mmol,79.7mg)、碳酸钾(2当量,2.61mmol,361mg,)、碳酸铯(0.3当量,0.39mmol,128mg)和碘化钠(6mg)。将混合物在ar下加热回流7天。在减压下蒸发溶剂,将残余物置于己烷/乙酸乙酯(大约10%)中,并用水和盐水洗涤。将有机层分离,经无水硫酸钠干燥,过滤并在减压下蒸发,获得油(1g)。将残余物(1g)在硅胶上通过重力柱色谱法(meoh于dcm中,0至4%)纯化。这产生呈无色油的化合物14(757mg 0.99mmol,76%)。
[0261]
实施例20
[0262]
化合物15的合成
[0263]
向2
‑
己基癸基5
‑
溴戊酸酯(2当量,1.22g,3mmol)的15ml无水thf(打开2个月)溶液中添加4
‑
氨基
‑1‑
丁醇(1当量,1.5mmol,0.134mg,139ul)、碳酸钾(2当量,3mmol,415mg)、碳酸铯(0.3当量,0.45mmol,146mg)和碘化钠(6mg)。将混合物在ar下加热回流6天。在减压下蒸发溶剂,将残余物置于己烷和乙酸乙酯(大约10%)的混合物中,并用水和盐水洗涤。将有机层分离,经无水硫酸钠干燥,过滤并在减压下蒸发,获得油(1.12g)。将残余物在硅胶上通过柱色谱法(meoh于氯仿中,0至5%)纯化。这产生呈无色油的化合物15(487mg,0.66mmol,44%)。1hnmr(400mhz,cdcl3)δ:5.99(s,1h),3.98(d,5.8hz,4h),3.56(t样,4.8hz,2h),2.48
‑
2.41(m,6h),2.33(t,7.4hz,4h),1.70
‑
1.57(m,10h),1.55
‑
1.47(m,4h),1.35
‑
1.21(48h),0.89(t样,6.8hz,12h)。
[0264]
实施例21
[0265]
化合物16的合成
[0266]
向3
‑
氨基
‑1‑
丙醇(0.37mmol,28mg)的无水乙腈(15ml)溶液中添加2
‑
己基癸基6
‑
溴己酸酯(1.9当量,294mg,0.7mmol)、n,n
‑
二异丙基乙胺(2当量,0.74mmol,96m)和碘化钠(5mg),并将混合物(两层)在59℃(油浴)下于耐压烧瓶中加热3天。浓缩混合物,将残余物置
于己烷和乙酸乙酯(大约5:1,100ml)的混合物中,用水、盐水洗涤,经硫酸钠干燥,过滤并浓缩。获得了略带黄色的油(大约300mg)。将粗产物(300mg)在硅胶上通过快速柱色谱法(meoh于氯仿中,0至4.4%)纯化。这产生呈无色油的化合物16(95mg,0.13mmol,36%)。1hnmr(400mhz,cdcl3)δ:5.61
‑
5.44(br.s,1h),3.97(d,5.8hz,4h),3.80(t样,5.1hz,2h),2.63(t样,5.6hz,2h),2.43
‑
2.39(m,4h),2.32(t,7.5hz,4h),1.70
‑
1.59(m,8h),1.55
‑
1.45(m,4h),1.36
‑
1.21(52h),0.89(t样,6.8hz,12h)。
[0267]
实施例22
[0268]
化合物17的合成
[0269]
向2
‑
己基癸基6
‑
溴己酸酯(2当量,1.32g,3.14mmol)的15ml无水thf溶液中添加4
‑
氨基
‑1‑
丁醇(1当量,1.57mmol,140mg,145ul)、碳酸钾(2当量,3.14mmol,434mg)、碳酸铯(0.3当量,0.47mmol,153mg)和碘化钠(6mg)。将混合物在75℃(油浴)下于ar中在耐压圆底烧瓶中加热6天。冷却并浓缩反应混合物。将残余物置于己烷和乙酸乙酯(大约9:1)的混合物中,用水、盐水洗涤,经硫酸钠干燥,过滤并浓缩至干(1.28g无色油)。将粗产物在硅胶上通过快速柱色谱法(meoh于氯仿中,0至5%)纯化。这产生呈无色油的化合物17(581mg,0.76mmol,48%)。1hnmr(400mhz,cdcl3)δ:6.43
‑
6.17(br.s,1h),3.97(d,5.8hz,4h),3.55(t样,4.7hz,2h),2.46
‑
2.40(m,6h),2.31(t,7.5hz,4h),1.70
‑
1.59(m,10h),1.55
‑
1.45(m,4h),1.36
‑
1.21(52h),0.89(t样,6.7hz,12h)。
[0270]
实施例23
[0271]
化合物20的合成
[0272]
向2
‑
己基癸基8
‑
溴辛酸酯(2当量,3.09g,6.9mmol)的30ml无水thf溶液中添加4
‑
氨基
‑1‑
丁醇(1当量,3.45mmol,308mg)、碳酸钾(2当量,6.9mmol,954mg)、碳酸铯(0.3当量,1.04mmol,337mg)和碘化钠(10mg)。将在耐压圆底烧瓶中的混合物在ar中在64
‑
70℃(油浴)下加热6天。冷却并浓缩混合物。将残余物置于己烷和乙酸乙酯(9:1)的混合物中,用水、盐水洗涤,经硫酸钠干燥,过滤并浓缩至干(无色油)。将粗产物在硅胶上通过快速干燥柱色谱法(meoh于氯仿中,0至4.2%)纯化。这产生呈无色油的化合物20(1.28g,1.56mmol,45%)。1hnmr(400mhz,cdcl3)δ:6.64
‑
6.45(br.s,1h),3.97(d,5.8hz,4h),3.62
‑
3.51(br.2h),3.07
‑
2.34(br.6h),2.30(t,7.5hz,4h),1.71
‑
1.40(m,14h),1.39
‑
1.19(m,60h),0.89(t样,6.8hz,12h)。
[0273]
实施例24
[0274]9‑
(2
’‑
乙基己酰基氧基)壬
‑1‑
醛的合成
[0275]
用2
‑
乙基己酸(9.0g)、dcc(14.3g)和dmap(9.1g)处理壬
‑
1,9
‑
二醇(10.1g)的二氯甲烷(150ml)溶液。搅拌溶液过夜。过滤反应混合物,并去除溶剂。将残余物悬浮于己烷中,并过滤。用稀盐酸洗涤滤液。将有机相经无水硫酸镁干燥,通过硅胶床过滤并去除溶剂。使用甲醇/二氯甲烷(0
‑
8%)梯度,使粗产物通过硅胶柱,产生呈油状物的9
‑
(2
’‑
乙基己酰基氧基)壬
‑1‑
醇(7.2g)。
[0276]
将9
‑
(2
’‑
乙基己酰基氧基)壬
‑1‑
醇溶于二氯甲烷(100ml)中,并用氯铬酸吡啶鎓盐(7.5g)处理一小时。添加己烷(400ml),并通过硅胶床过滤上清液。从滤液中去除溶剂,并将所得的油溶于己烷中。通过硅胶床过滤悬浮液并去除溶剂,产生呈无色油的9
‑
(2
’‑
乙基己酰基氧基)壬
‑1‑
醛(6g)。
[0277]
实施例25
[0278]9‑
(2
’‑
丁基辛酰基氧基)壬
‑1‑
醛的合成
[0279]
用2
‑
丁基辛酸(5.0g)、dcc(7.7g)和dmap(4.5g)处理壬
‑
1,9
‑
二醇(12.0g)的二氯甲烷(150ml)溶液。搅拌溶液过夜。过滤反应混合物,并去除溶剂。将残余物悬浮于己烷中,并过滤。用稀盐酸洗涤滤液。将有机相经无水硫酸镁干燥,通过硅胶床过滤,并去除溶剂。使用甲醇/二氯甲烷(0
‑
4%)梯度,使粗产物通过硅胶柱,产生呈油状物的9
‑
(2
’‑
丁基辛酰基氧基)壬
‑1‑
醇(6g)。
[0280]
将9
‑
(2
’‑
丁基辛酰基氧基)壬
‑1‑
醇溶于二氯甲烷(100ml)中,并用氯铬酸吡啶鎓盐(3.8g)处理过夜。添加己烷(300ml),并通过硅胶床过滤上清液。从滤液中去除溶剂,并将所得的油溶于己烷中。通过硅胶床过滤悬浮液并去除溶剂,产生呈无色油的9
‑
(2
’‑
丁基辛酰基氧基)壬
‑1‑
醛(3.1g)。
[0281]
实施例26
[0282]6‑
(2
’‑
丁基辛酰基氧基)己
‑1‑
醛的合成
[0283]
用2
‑
丁基辛酸(5.0g)、dcc(7.6g)和dmap(4.8g)处理己
‑
1,6
‑
二醇(9.4g)的二氯甲烷(150ml)溶液。搅拌溶液过夜。过滤反应混合物,并去除溶剂。将残余物悬浮于己烷中,并过滤。用稀盐酸洗涤滤液。将有机相经无水硫酸镁干燥,通过硅胶床过滤并去除溶剂。使用甲醇/二氯甲烷(0
‑
4%)梯度,使粗产物通过硅胶柱,产生呈油状物的6
‑
(2
’‑
丁基辛酰基氧基)己
‑1‑
醇(4.5g)。
[0284]
将6
‑
(2
’‑
丁基辛酰基氧基)己
‑1‑
醇溶于二氯甲烷(100ml)中,并用氯铬酸吡啶鎓盐(4.8g)处理两小时。添加己烷(300ml),并通过硅胶床过滤上清液。从滤液中去除溶剂,并将所得的油溶于己烷中。通过硅胶床过滤悬浮液并去除溶剂,产生呈无色油的6
‑
(2
’‑
丁基辛酰基氧基)己
‑1‑
醛(3.9g)。
[0285]
实施例27
[0286]6‑
(2
’‑
辛基十二烷酰基氧基)己
‑1‑
醛的合成
[0287]
用2
‑
辛基十二烷酸(9.9g)、dcc(7.5g)和dmap(4.7g)处理己
‑
1,6
‑
二醇(11.5g)的二氯甲烷(150ml)/thf(20ml)溶液。搅拌溶液过夜。过滤反应混合物,并去除溶剂。将残余物悬浮于己烷中,并过滤。用稀盐酸洗涤滤液。将有机相经无水硫酸镁干燥,通过硅胶床过滤,并去除溶剂。使用甲醇/二氯甲烷(0
‑
4%)梯度,使粗产物通过硅胶柱,产生呈油状物的6
‑
(2
’‑
辛基十二烷酰基氧基)己
‑1‑
醇(7.4g)。
[0288]
将6
‑
(2
’‑
辛基十二烷酰基氧基)己
‑1‑
醇溶于二氯甲烷(100ml)中,并用氯铬酸吡啶鎓盐(4.0g)处理两小时。添加乙醚(300ml),并通过硅胶床过滤上清液。从滤液中去除溶剂,并将所得的油溶于己烷中。通过硅胶床过滤悬浮液并去除溶剂,产生呈无色油的6
‑
(2
’‑
辛基十二烷酰基氧基)己
‑1‑
醛(5.3g)。
[0289]
实施例28
[0290]6‑
(2
’‑
癸基十四烷酰基氧基)己
‑1‑
醛的合成
[0291]
用2
‑
癸基十四烷酸(6.1g)、dcc(4.9g)和dmap(3.1g)处理己
‑
1,6
‑
二醇(9.6g)的二氯甲烷(150ml)溶液。搅拌溶液过夜。过滤反应混合物,并去除溶剂。将残余物悬浮于己烷中,并过滤。用稀盐酸洗涤滤液。将有机相经无水硫酸镁干燥,通过硅胶床过滤,并去除溶剂。使用甲醇/二氯甲烷(0
‑
4%)梯度,使粗产物通过硅胶柱,产生6
‑
(2
’‑
癸基十四烷酰基氧
基)己
‑1‑
醇(4.6g)。
[0292]
将6
‑
(2
’‑
癸基十四烷酰基氧基)己
‑1‑
醇溶于二氯甲烷(100ml)中,并用氯铬酸吡啶鎓盐(3.2g)处理两小时。添加己烷(300ml),并通过硅胶床过滤上清液。从滤液中去除溶剂,并将所得产物溶于己烷中。通过硅胶床过滤悬浮液并去除溶剂,产生6
‑
(2
’‑
癸基十四烷酰基氧基)己
‑1‑
醛(4.2g)。
[0293]
实施例29
[0294]
12
‑
(2
’‑
己基癸酰基氧基)十二
‑1‑
醛的合成
[0295]
用2
‑
己基癸酸(10.6g)、dcc(10.2g)和dmap(7.5g)处理十二烷
‑
1,12
‑
二醇(25.0g)的二氯甲烷(300ml)/thf(100ml)溶液。搅拌溶液过夜。过滤反应混合物,并去除溶剂。将残余物悬浮于己烷中,并过滤。用水洗涤滤液。将有机相经无水硫酸镁干燥,通过硅胶床过滤,并去除溶剂。使用己烷随后使用二氯甲烷,使粗产物通过硅胶柱,产生呈油状物的12
‑
(2
’‑
己基癸酰基氧基)十二烷
‑1‑
醇(7.9g)。
[0296]
将12
‑
(2
’‑
己基癸酰基氧基)十二烷
‑1‑
醇溶于二氯甲烷(150ml)中,并用氯铬酸吡啶鎓盐(4.0g)处理三小时。添加己烷(300ml),并通过硅胶床过滤上清液。从滤液中去除溶剂,并将所得的油溶于己烷中。通过硅胶床过滤悬浮液并去除溶剂,产生呈无色油的12
‑
(2
’‑
己基癸酰基氧基)十二
‑1‑
醛(3.9g)。
[0297]
实施例30
[0298]9‑
(2
’‑
己基癸酰基氧基)壬
‑1‑
醛的合成
[0299]
用2
‑
己基癸酸(25.0g)、dcc(22.0g)和dmap(15.0g)处理壬
‑
1,9
‑
二醇(46.8g)的二氯甲烷(600ml)溶液。搅拌溶液过夜。过滤反应混合物,并去除溶剂。将残余物悬浮于己烷中,并过滤。用稀盐酸洗涤滤液。将有机相经无水硫酸镁干燥,通过硅胶床过滤,并去除溶剂。使用己烷随后使用甲醇/二氯甲烷(0
‑
8%)梯度,使粗产物通过硅胶柱,产生呈油状物的9
‑
(2
’‑
己基癸酰基氧基)壬
‑1‑
醇(22g)。
[0300]
将9
‑
(2
’‑
己基癸酰基氧基)壬
‑1‑
醇(5.0g)溶于二氯甲烷(50ml)中,并用氯铬酸吡啶鎓盐(2.7g)处理一小时。添加己烷(200ml),并通过硅胶床过滤上清液。从滤液中去除溶剂,并将所得的油溶于己烷中。通过硅胶床过滤悬浮液并去除溶剂,产生呈无色油的9
‑
(2
’‑
己基癸酰基氧基)壬
‑1‑
醛(3.6g)。
[0301]
实施例31
[0302]
化合物22的合成
[0303]
将9
‑
(2
’‑
己基癸酰基氧基)壬
‑1‑
醛(2.2g)、乙酸(0.15g)和4
‑
氨基丁
‑1‑
醇(0.20g)的二氯甲烷(20ml)溶液用三乙酰氧基硼氢化钠(1.30g)处理过夜。用碳酸氢钠水溶液洗涤该溶液。将有机相经无水硫酸镁干燥、过滤并去除溶剂。使用乙酸/甲醇/二氯甲烷(2
‑
0/0
‑
12/98
‑
88%)梯度,使残余物通过硅胶柱。用碳酸氢钠水溶液洗涤纯的级分,产生呈无色油的化合物22(0.93g)。
[0304]
实施例32
[0305]
化合物23的合成
[0306]
将12
‑
(2
’‑
己基癸酰基氧基)十二
‑1‑
醛(2.0g)、乙酸(0.09g)和4
‑
氨基丁
‑1‑
醇(0.14g)的二氯甲烷(20ml)溶液用三乙酰氧基硼氢化钠(0.71g)处理过夜。用碳酸氢钠水溶液洗涤该溶液。将有机相经无水硫酸镁干燥、过滤并去除溶剂。使用乙酸/甲醇/二氯甲烷
(2
‑
0/0
‑
12/98
‑
88%)梯度,使残余物通过硅胶柱。用碳酸氢钠水溶液洗涤纯的级分,产生呈无色油的化合物23(1.0g)。
[0307]
实施例33
[0308]
化合物24的合成
[0309]
将9
‑
(2
’‑
乙基己酰基氧基)壬
‑1‑
醛(3.0g)、乙酸(0.11g)和4
‑
氨基丁
‑1‑
醇(0.17g)的二氯甲烷(50ml)溶液用三乙酰氧基硼氢化钠(0.89g)处理过夜。用碳酸氢钠水溶液洗涤该溶液。将有机相经无水硫酸镁干燥、过滤并去除溶剂。使用乙酸/甲醇/二氯甲烷(2
‑
0/0
‑
10/98
‑
90%)梯度,使残余物通过硅胶柱。用碳酸氢钠水溶液洗涤纯的级分,产生呈无色油的化合物24(0.69g)。
[0310]
实施例34
[0311]
化合物25的合成
[0312]
将9
‑
(2
’‑
丁基辛酰基氧基)壬
‑1‑
醛(2.6g)、乙酸(0.20g)和4
‑
氨基丁
‑1‑
醇(0.26g)的二氯甲烷(50ml)溶液用三乙酰氧基硼氢化钠(1.42g)处理过夜。用碳酸氢钠水溶液洗涤该溶液。将有机相经无水硫酸镁干燥、过滤并去除溶剂。使用乙酸/甲醇/二氯甲烷(2
‑
0/0
‑
12/98
‑
88%)梯度,使残余物通过硅胶柱。用碳酸氢钠水溶液洗涤纯的级分,产生呈无色油的化合物25(0.82g)。
[0313]
实施例35
[0314]
化合物26的合成
[0315]
将6
‑
(2
’‑
辛基十二烷酰基氧基)己
‑1‑
醛(2.7g)、乙酸(0.20g)和4
‑
氨基丁
‑1‑
醇(0.20g)的二氯甲烷(20ml)溶液用三乙酰氧基硼氢化钠(1.30g)处理过夜。用碳酸氢钠水溶液洗涤该溶液。将有机相经无水硫酸镁干燥、过滤并去除溶剂。使用乙酸/甲醇/二氯甲烷(2
‑
0/0
‑
12/98
‑
88%)梯度,使残余物通过硅胶柱。用碳酸氢钠水溶液洗涤纯的级分,产生呈无色油的化合物26(0.21g)。
[0316]
实施例36
[0317]
化合物27的合成
[0318]
将6
‑
(2
’‑
癸基十四烷酰基氧基)己
‑1‑
醛(2.1g)、乙酸(0.11g)和4
‑
氨基丁
‑1‑
醇(0.13g)的二氯甲烷(30ml)溶液用三乙酰氧基硼氢化钠(0.70g)处理过夜。用碳酸氢钠水溶液洗涤该溶液。将有机相经无水硫酸镁干燥、过滤并去除溶剂。使用乙酸/甲醇/二氯甲烷(2
‑
0/0
‑
12/98
‑
88%)梯度,使残余物通过硅胶柱。用碳酸氢钠水溶液洗涤纯的级分,产生呈无色油的化合物27(0.90g)。
[0319]
实施例37
[0320]
化合物28的合成
[0321]
将6
‑
(2
’‑
丁基辛酰基氧基)己
‑1‑
醛(2.0g)、乙酸(0.13g)和3
‑
氨基丙
‑1‑
醇(0.13g)的二氯甲烷(20ml)溶液用三乙酰氧基硼氢化钠(1.0g)处理过夜。用碳酸氢钠水溶液洗涤该溶液。将有机相经无水硫酸镁干燥、过滤并去除溶剂。使用乙酸/甲醇/二氯甲烷(2
‑
0/0
‑
8/98
‑
92%)梯度,使残余物通过硅胶柱。用碳酸氢钠水溶液洗涤纯的级分,产生呈无色油的化合物28(0.77g)。
[0322]
实施例38
[0323]
化合物30的合成
[0324]
将6
‑
(2
’‑
己基癸酰基氧基)己
‑1‑
醛(2.4g)、乙酸(0.15g)和3
‑
氨基丙
‑
1,2
‑
二醇(0.21g)的二氯甲烷(20ml)溶液用三乙酰氧基硼氢化钠(1.76g)处理过夜。用碳酸氢钠水溶液洗涤该溶液。将有机相经无水硫酸镁干燥、过滤并去除溶剂。使用乙酸/甲醇/二氯甲烷(2
‑
0/0
‑
12/98
‑
88%)梯度,使残余物通过硅胶柱。用碳酸氢钠水溶液洗涤纯的级分,产生呈无色油的化合物30(0.60g)。
[0325]
实施例39
[0326]
化合物31的合成
[0327]
将6
‑
(2
’‑
己基癸酰基氧基)己
‑1‑
醛(2.4g)、乙酸(0.15g)和2
‑
氨基丁
‑1‑
醇(0.20g)的二氯甲烷(20ml)溶液用三乙酰氧基硼氢化钠(1.1g)处理两小时。用稀氢氧化钠水溶液洗涤该溶液。将有机相经无水硫酸镁干燥、过滤并去除溶剂。使用乙酸/甲醇/二氯甲烷(2
‑
0/0
‑
4/98
‑
96%)梯度,使残余物通过硅胶柱。用碳酸氢钠水溶液洗涤纯的级分,产生呈无色油的化合物31(0.31g)。
[0328]
实施例40
[0329]
化合物37的合成
[0330]
将6
‑
(2
’‑
辛基十二烷酰基氧基)己
‑1‑
醛(2.7g)、乙酸(0.20g)和3
‑
氨基丙
‑1‑
醇(0.17g)的二氯甲烷(20ml)溶液用三乙酰氧基硼氢化钠(1.3g)处理过夜。用碳酸氢钠水溶液洗涤该溶液。将有机相经无水硫酸镁干燥、过滤并去除溶剂。使用乙酸/甲醇/二氯甲烷(2
‑
0/0
‑
12/98
‑
88%)梯度,使残余物通过硅胶柱,用碳酸氢钠水溶液洗涤纯的级分,产生呈无色油的化合物37(0.22g)。
[0331]
实施例41
[0332]
化合物38的合成
[0333]
将12
‑
(2
’‑
己基癸酰基氧基)十二
‑1‑
醛(1.8g)、乙酸(0.08g)和3
‑
氨基丙
‑1‑
醇(0.11g)的二氯甲烷(10ml)溶液用三乙酰氧基硼氢化钠(0.64g)处理过夜。用碳酸氢钠水溶液洗涤该溶液。将有机相经无水硫酸镁干燥、过滤并去除溶剂。使用乙酸/甲醇/二氯甲烷(2
‑
0/0
‑
10/98
‑
90%)梯度,使残余物通过硅胶柱。用碳酸氢钠水溶液洗涤纯的级分,产生呈无色油的化合物38(0.83g)。
[0334]
实施例42
[0335]
化合物39的合成
[0336]
将4
‑
氨基丁酸乙酯盐酸盐(1.28mmol,214mg)、2
‑
己基癸基6
‑
溴己酸酯(1.9当量,2.43mmol,1.02g)、n,n
‑
二异丙基乙胺(3.5当量,4.48mmol,579mg)和碘化钠(5mg)在无水乙腈(15ml)中的混合物(两层)在60℃下于耐压烧瓶中加热2天。冷却并浓缩混合物。将残余物置于己烷和乙酸乙酯(大约5:1,100ml)的混合物中,用水、盐水洗涤,经硫酸钠干燥,过滤并浓缩。获得棕色油(大约1.04g)。将粗产物在硅胶上通过快速柱色谱法(meoh于dcm中,0至3.5%)纯化。这产生呈无色油的化合物39(334mg,0.41mmol,43%)。1hnmr(400mhz,cdcl3)δ:4.13(q,7.1hz,2h),3.97(d,5.8hz,4h),2.43
‑
2.34(m,6h),2.33
‑
2.28(m,6h),1.73(五重峰,7.3hz,2h),1.68
‑
1.58(m,6h),1.47
‑
1.37(m,4h),1.36
‑
1.20(54h),0.89(t样,6.8hz,12h)。
[0337]
实施例43
[0338]
化合物40的合成
[0339]
将6
‑
(2
’‑
己基癸酰基氧基)己
‑1‑
醛(2.4g)、乙酸(0.15g)和1
‑
氨基丁
‑2‑
醇(0.10g)的二氯甲烷(20ml)溶液用三乙酰氧基硼氢化钠(1.8g)处理两小时。用碳酸氢钠水溶液洗涤该溶液。将有机相经无水硫酸镁干燥、过滤并去除溶剂。使用乙酸/甲醇/二氯甲烷(2
‑
0/0
‑
8/98
‑
92%)梯度,使残余物通过硅胶柱。用碳酸氢钠水溶液洗涤纯的级分,产生呈无色油的化合物40(0.85g)。
[0340]
实施例44
[0341]
化合物41的合成
[0342]
将6
‑
(2
’‑
己基癸酰基氧基)己
‑1‑
醛(2.4g)、乙酸(0.19g)和3
‑
甲氧基丙胺(0.21g)的二氯甲烷(20ml)溶液用三乙酰氧基硼氢化钠(1.8g)处理过夜。用碳酸氢钠水溶液洗涤该溶液。将有机相经无水硫酸镁干燥、过滤并去除溶剂。使用乙酸/甲醇/二氯甲烷(2
‑
0/0
‑
8/98
‑
92%)梯度,使残余物通过硅胶柱。用碳酸氢钠水溶液洗涤纯的级分,产生呈无色油的化合物41(0.77g)。
[0343]
实施例45
[0344]
化合物42的合成
[0345]
将6
‑
(2
’‑
丁基辛酰基氧基)己
‑1‑
醛(2.0g)、乙酸(0.13g)和4
‑
氨基丁
‑1‑
醇(0.20g)的二氯甲烷(20ml)溶液用三乙酰氧基硼氢化钠(1.03g)处理过夜。用碳酸氢钠水溶液洗涤该溶液。将有机相经无水硫酸镁干燥、过滤并去除溶剂。使用乙酸/甲醇/二氯甲烷(2
‑
0/0
‑
8/98
‑
92%)梯度,使残余物通过硅胶柱。用碳酸氢钠水溶液洗涤纯的级分,产生呈无色油的化合物42(0.54g)。
[0346]
实施例46
[0347]
化合物43的合成
[0348]
将9
‑
(2
’‑
乙基己酰基氧基)壬
‑1‑
醛(3.0g)、乙酸(0.11g)和3
‑
氨基丙
‑1‑
醇(0.14g)的二氯甲烷(50ml)溶液用三乙酰氧基硼氢化钠(0.91g)处理过夜。用碳酸氢钠水溶液洗涤该溶液。将有机相经无水硫酸镁干燥、过滤并去除溶剂。使用乙酸/甲醇/二氯甲烷(2
‑
0/0
‑
6/98
‑
94%)梯度,使残余物通过硅胶柱。用碳酸氢钠水溶液洗涤纯的级分,产生呈无色油的化合物43(1.01g)。
[0349]
实施例47
[0350]
化合物44的合成
[0351]
将6
‑
(2
’‑
癸基十四烷酰基氧基)己
‑1‑
醛(2.1g)、乙酸(0.11g)和3
‑
氨基丙
‑1‑
醇(0.11g)的二氯甲烷(30ml)溶液用三乙酰氧基硼氢化钠(0.71g)处理过夜。用碳酸氢钠水溶液洗涤该溶液。将有机相经无水硫酸镁干燥、过滤并去除溶剂。使用乙酸/甲醇/二氯甲烷(2
‑
0/0
‑
8/98
‑
96%)梯度,使残余物通过硅胶柱。用碳酸氢钠水溶液洗涤纯的级分,产生呈无色油的化合物44(1.07g)。
[0352]
实施例48
[0353]
化合物45的合成
[0354]
将9
‑
(2
’‑
丁基辛酰基氧基)壬
‑1‑
醛(2.6g)、乙酸(0.17g)和3
‑
氨基丙
‑1‑
醇(0.21g)的二氯甲烷(50ml)溶液用三乙酰氧基硼氢化钠(1.34g)处理过夜。用碳酸氢钠水溶液洗涤该溶液。将有机相经无水硫酸镁干燥、过滤并去除溶剂。使用乙酸/甲醇/二氯甲烷(2
‑
0/0
‑
8/98
‑
96%)梯度,使残余物通过硅胶柱。用碳酸氢钠水溶液洗涤纯的级分,产生呈
无色油的化合物45(1.1g)。
[0355]
实施例49
[0356]
化合物46的合成
[0357]
向2
‑
氨基乙醇(96.5mg,1.58mmol,95.4ul,mw 61.08,d 1.012)的15ml 2
‑
丙醇溶液中添加2
‑
己基癸基8
‑
溴辛酸酯(1.8当量,1.27g,2.84mmol)、碳酸钾(1.9当量,3mmol,414mg)、碳酸铯(0.3当量,0.47mmol,154mg)和碘化钠(10mg),并加热3天(油浴60℃)。浓缩混合物,并将残余物置于thf(10ml)中。向该混合物中添加更多的氨基乙醇(80mg,1.3mmol)。在70℃下再继续加热3天。在总共6天之后,冷却、过滤并浓缩反应混合物。将残余物在硅胶上通过快速干柱色谱法(甲醇于氯仿中,1%至4.2%)纯化。这产生呈无色油的化合物46(334mg,0.42mmol,30%)。1hnmr(400mhz,cdcl3)δ:4.09
‑
4.06(m,2h),3.97(d,5.8hz,4h),3.39
‑
3.36(m,2h),3.31
‑
3.23(m,4h),2.31(t,7.5hz,4h),1.88
‑
1.56(m,12h),1.43
‑
1.19(59h),0.89(t样,6.8hz,12h)。
[0358]
实施例50
[0359]
化合物47的合成
[0360]
将6
‑
(2
’‑
己基癸酰基氧基)己
‑1‑
醛(3.0g)、乙酸(0.20g)和3
‑
氨基丙腈(0.21g)的二氯甲烷(30ml)溶液用三乙酰氧基硼氢化钠(1.3g)处理过夜。用碳酸氢钠水溶液洗涤该溶液。将有机相经无水硫酸镁干燥、过滤并去除溶剂。使用乙酸/甲醇/二氯甲烷(2
‑
0/0
‑
6/98
‑
94%)梯度,使残余物通过硅胶柱。用碳酸氢钠水溶液洗涤纯的级分,产生呈无色油的化合物47(0.29g)。
[0361]
实施例51
[0362]
化合物48的合成
[0363]
将6
‑
(2
’‑
己基癸酰基氧基)己
‑1‑
醛(3.0g)和4
‑
氨基丁酸乙酯盐酸盐(0.46g)的二氯甲烷(30ml)溶液用三乙酰氧基硼氢化钠(1.4g)处理过夜。用碳酸氢钠水溶液洗涤该溶液。将有机相经无水硫酸镁干燥、过滤并去除溶剂。使用乙酸/甲醇/二氯甲烷(2
‑
0/0
‑
8/98
‑
92%)梯度,使残余物通过硅胶柱。用碳酸氢钠水溶液洗涤纯的级分,产生呈无色油的化合物48(0.80g)。
[0364]
实施例52
[0365]
化合物49的合成
[0366]
向2
‑
丁基辛基8
‑
溴辛酸酯(2当量,1.877g,4.8mmol)的20ml无水thf溶液中添加4
‑
氨基
‑1‑
丁醇(1等量,2.4mmol,214mg,221ul)、碳酸钾(2当量,4.8mmol,664mg)、碳酸铯(0.3当量,0.72mmol,234mg)和碘化钠(大约5mg)。将在耐压圆底烧瓶中的混合物加热(油浴,80℃)6天。冷却并浓缩反应混合物。将残余物置于己烷和乙酸乙酯(大约5:1)的混合物中,用水、盐水洗涤,经硫酸钠干燥,过滤并浓缩。将残余物在硅胶上通过快速柱色谱法(甲醇于氯仿中,1至4%)纯化。这产生呈无色油的化合物49(857mg,1.21mmol,50%)。1hnmr(400mhz,cdcl3)δ:6.55(br.s,1h),3.97(d,5.8hz,4h),3.55(not well resolved triplet,2h),2.45
‑
2.40(m.6h),2.30(t,7.5hz,4h),1.71
‑
1.58(m,10h),1.51
‑
1.42(m,4h),1.39
‑
1.19(m,44h),0.93
‑
0.87(m,12h)。
[0367]
可以合并上述各种实施方案以提供其它实施方案。在与本文的具体教导和定义不一致的范围内,2015年10月28日提交的美国专利申请第62/247,616号,2016年4月27日提交
的美国专利申请第62/328,244号;本说明书中引用的和/或列于申请数据单中的所有美国专利、美国专利申请公开、美国专利申请、外国专利、外国专利申请和非专利出版物均通过引用整体并入本文。如果有必要,可使用各种专利、申请和出版物的概念修改各实施方案的方面,来提供其他的实施方案。根据上文详细的描述,可以对实施方案进行这些和其他的改变。通常,在权利要求中,不应将所使用的术语解释为将权利要求限定于本说明书和权利要求中公开的特定实施方案,而是应当将权利要求解释为包括所有可能的实施方案,以及这些权利要求有权享有的等价物的全部范围。因此,权利要求不受本公开内容限定。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1