一类高效等规聚丙烯β晶型成核剂及其制备方法和应用
一类高效等规聚丙烯
β
晶型成核剂及其制备方法和应用
技术领域
1.本发明属于聚丙烯研究技术领域,具体地说,涉及一类高效等规聚丙烯β晶型成核剂及其制备方法和应用。
背景技术:
2.自1957年实现工业化生产以来,聚丙烯已成为发展速度最快,应用范围最广的通用塑料之一。聚丙烯具有良好的机械性能、加工性能以及耐热性能,生产成本低,用于生产注塑制品、薄膜、管材和纤维等制品,在汽车、家电、包装、电池隔膜等领域中得到了广泛的应用。
3.通过调控分子链结构和冷却条件,以及加入成核剂可使等规聚丙烯形成α、β、γ晶和一种中介相。在这几种晶型中,等规聚丙烯的分子链均采取31螺旋构象的方式排入晶格。其中,热力学最稳定的α晶在通常的加工条件下可以形成,以α晶为主的等规聚丙烯具有较高的屈服强度和模量。β晶处于热力学亚稳态,在剪切作用、高降温梯度及添加β晶型成核剂的条件下可以形成。加入β晶型成核剂是目前工业应用中获得高β晶含量等规聚丙烯最有效的方法。
4.β晶型成核剂能够显著提高等规聚丙烯制品的韧性(抗冲击性能和断裂伸长率)、热变形温度,通过拉伸形变获得高微孔率,拓展了等规聚丙烯的应用领域。高效的β晶型成核剂主要包括以下几类:1)具有准平面结构的稠环芳烃化合物,如γ
‑
喹吖啶酮等;2)二元羧酸和某些ⅱa族金属的氧化物、氢氧化物或盐组成的复合物及ⅱa族金属盐,如庚二酸/硬脂酸钙体系、己二酸锌、四氢邻苯二甲酸钡等;3)酰胺类化合物,如n,n
′‑
二环己基对苯二甲酰胺(dcht)和n,n
′‑
二环己基
‑
2,6
‑
萘二甲酰胺(dchn)等;4)稀土类化合物,如wbg;5)(主链及侧链型)液晶聚合物(如pbdps和lcp
‑
na2)以及生物基聚合物(如聚多巴胺)等。
5.1967年leugering首次发现γ
‑
喹吖啶酮是一种高效的β晶型成核剂。oliveira等发现δ
‑
喹吖啶酮也能诱导形成高β晶含量的等规聚丙烯。li等发现一些其它的有机颜料也具有较好的β晶成核效果。然而,这类稠环芳烃化合物成本高、有颜色,会影响等规聚丙烯产品的外观和透明性。
6.us05231126a、ep0682066b2、us20100010168a1、de3610644a1等公开了一系列由二元羧酸和ⅱa族金属的氧化物、氢氧化物或盐组成的复合物,能够诱导等规聚丙烯形成大量甚至纯的β晶,但这类β晶型成核剂热稳定性差,在加工过程中容易析出,影响β晶成核效率及等规聚丙烯产品的力学性能。cn1966563a和cn102181092a分别公开了一种环状二羧酸盐类化合物和四氢苯酐的羧酸金属盐可作为等规聚丙烯β晶型成核剂,β晶成核效率较高,但成本较高。
7.cn1114651c和cn101265342b公开了一种稀土配合物类β晶型成核剂,由多组分化合物组合而成,结构不明确,成分复杂。
8.近年来有研究报道(主链及侧链型)液晶聚合物也具有诱导等规聚丙烯形成β晶型的能力。胡建设等发现侧链型液晶聚合物lcp
‑
na2添加量为1.0wt%时,β晶型的相对含量为
70%。li等发现主链型液晶聚合物pbdps是一种高效的β晶型成核剂,添加量为4.0wt%时,β晶型的相对含量可达到96.6%,但添加量高,合成困难,成本较高。cn105255010a公开了一种成核效率较高,与聚丙烯基体相容性好的β晶型成核剂
‑
聚多巴胺,当成核剂的添加量为0.5wt%时,xrd和dsc测定等规聚丙烯中β晶型的相对含量最高,分别为69.8%和75.4%,成核效率一般。
9.ep0962489a2公开了一系列酰胺类化合物(如dcht和dchn等),这类化合物与聚丙烯相容性好,热稳定性好,β晶成核效率较高,是综合性能优良的β晶型成核剂,但品种较少。因此,研究与开发更多品种的酰胺类β晶型成核剂受到了研究人员的广泛关注。
10.有鉴于此特提出本发明。
技术实现要素:
11.本发明要解决的技术问题在于克服现有技术的不足,提供一类高效等规聚丙烯β晶型成核剂及其制备方法和应用。
12.为解决上述技术问题,本发明采用技术方案的基本构思是:
13.本发明第一目的提供了一类高效等规聚丙烯β晶型成核剂,具有如下结构通式:
14.r1
‑
x1
‑
a
‑
x2
‑
r2
15.其中,a为取代或未取代的c1‑
12
亚烷基、c5‑
12
亚环烷基、c2‑
12
亚烯基、c5‑
12
亚环烯基、亚苯基、亚萘基、亚联苯基中的一种;
16.‑
x1
‑
、
‑
x2
‑
分别为
‑
co
‑
nh
‑
和/或
‑
nh
‑
co
‑
;
17.r1和r2不同,分别独立地选自取代或未取代的c1‑
12
烷基、c5‑
12
环烷基、c5‑
12
环烯基、苯基、c5‑
12
杂环基中的一种。
18.进一步地,所述杂环基包括含n、o或s原子中的一个或多个。
19.在上述进一步的方案中,所述
‑
x1
‑
、
‑
x2
‑
中的碳原子均与所述a键合;或者所述
‑
x1
‑
、
‑
x2
‑
中的碳原子分别与所述r1和r2键合;或者所述
‑
x1
‑
、
‑
x2
‑
中的碳原子分别与所述a和所述r1/r2键合。
20.进一步地,所述
‑
x1
‑
、
‑
x2
‑
中的碳原子均与所述a键合,或者所述
‑
x1
‑
、
‑
x2
‑
中的碳原子分别与所述r1和r2键合。
21.进一步地,所述a任选被一个或多个以下取代基取代:c1‑
12
烷基、c1‑
12
烷氧基、c1‑
12
烷硫基、c1‑
12
烷基氨基、c1‑
12
烷基磺酰基、c1‑
12
烷基羰基、c1‑
12
烷氧基羰基、醛基、羧基、氰基、氨基、硝基、羟基、巯基、磺基和卤素;
22.所述r1和r2任选被一个或多个以下取代基取代:c1‑
12
烷基、c1‑
12
烷氧基、c1‑
12
烷硫基、c1‑
12
烷基氨基、c1‑
12
烷基磺酰基、c1‑
12
烷基羰基、c1‑
12
烷氧基羰基、醛基、羧基、氰基、氨基、硝基、巯基、磺基和卤素。
23.需要说明的是,上述a、r1和r2中取代基的情况并不限于此,包含其他取代基构成的具有上述结构通式的等规聚丙烯β晶型成核剂均属于本发明的保护范围。
24.作为本发明的一种实施方案,一类高效等规聚丙烯β晶型成核剂,具有如下结构式(i):
[0025][0026]
作为本发明的另一种实施方案,一类所述高效等规聚丙烯β晶型成核剂,具有如下结构式(ii):
[0027][0028]
作为本发明的再一种实施方案,一类所述高效等规聚丙烯β晶型成核剂,具有如下结构式(iii):
[0029][0030]
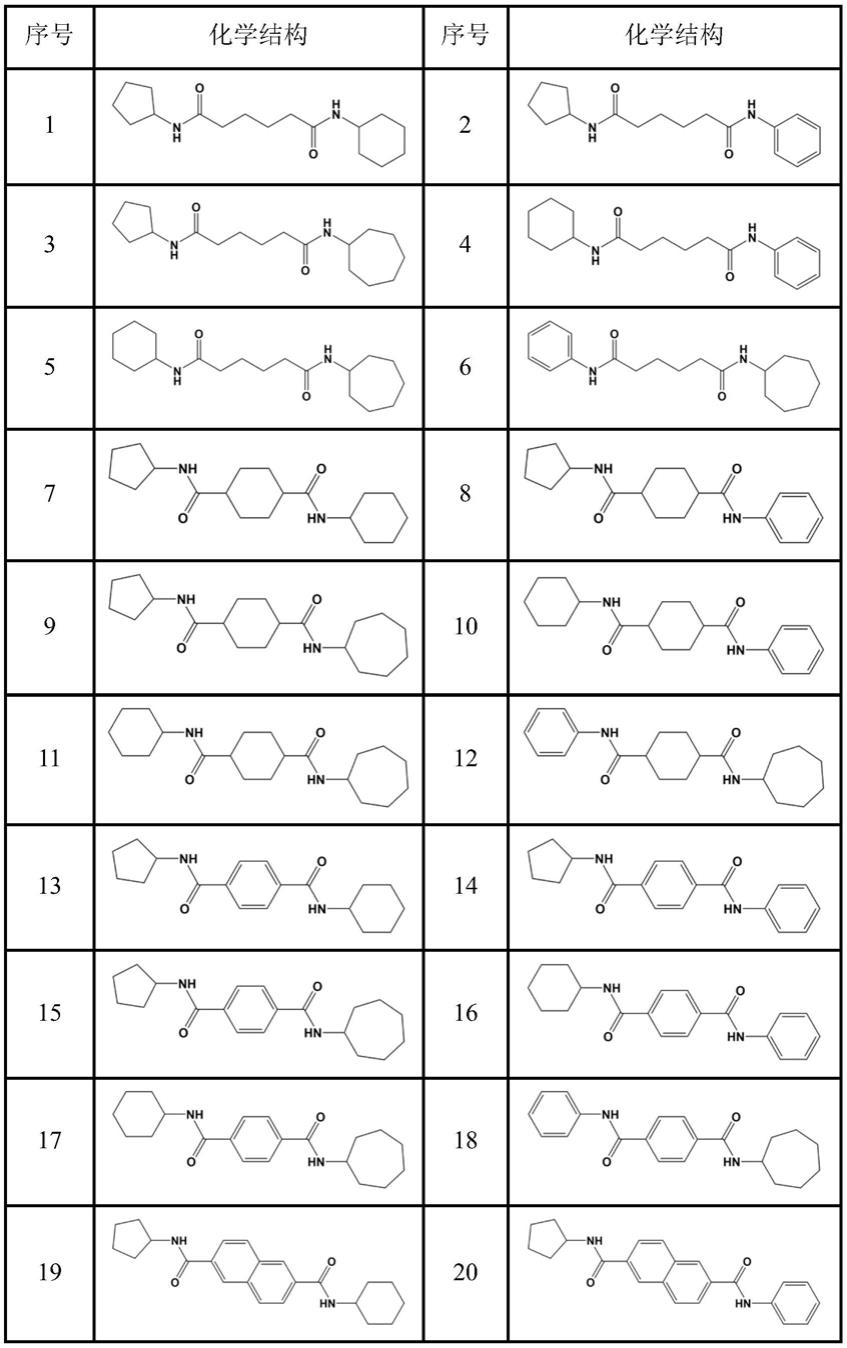
表1示出了式(i)中不对称取代二酰胺类化合物化学结构的非限制性实例。
[0031]
表1
[0032]
[0033][0034]
表2示出了式(ii)中不对称取代甲酰胺类化合物化学结构的非限制性实例。
[0035]
表2
[0036]
[0037]
[0038][0039]
表3示出了式(iii)中不对称取代酰胺类化合物化学结构的非限制性实例。
[0040]
表3
[0041]
[0042]
[0043]
[0044][0045]
本发明另一目的提供了一类高效等规聚丙烯β晶型成核剂的制备方法,包括如下步骤:
[0046]
(1)将r1—nh2和催化剂在有机溶剂中常温下进行反应后,经萃取、旋蒸,制得
[0047]
(2)向中加入氢氧化钠、水和无水乙醇进行水解反应后,经旋蒸、酸化、抽滤,制得
[0048]
(3)将r2—nh2和催化剂在有机溶剂中常温下进行反应后,经旋蒸、抽滤、淋洗、重结晶后,制得
[0049]
本发明另一目的提供了一类高效等规聚丙烯β晶型成核剂的制备方法,包括如下步骤:
[0050]
(1)将2on—a—nh2、r1—cooh和催化剂在有机溶剂中常温下进行反应后,经萃取、旋蒸,制得
[0051]
(2)和催化剂在有机溶剂中进行还原反应后,经冷却、旋蒸、萃取、干燥后,制得
[0052]
(3)将r2—cooh和催化剂在有机溶剂中常温下进行反应后,经旋蒸、抽滤、淋洗、重结晶后,制得
[0053]
本发明另一目的提供了一类高效等规聚丙烯β晶型成核剂的制备方法,包括如下步骤:
[0054]
(1)r2—nh2和催化剂在有机溶剂中常温下进行反应后,经萃取、旋蒸,制得
[0055]
(2)将r1—cooh和催化剂在有机溶剂中常温下进行反应后,经旋蒸、抽滤、淋洗、重结晶后,制得
[0056]
现有技术中对称取代酰胺类β晶型成核剂在制备过程中是将环烷胺、对苯二甲酸或对苯二甲酰氯、吡啶和二氯甲烷一起添加到容器中进行反应,即可制备得到对称取代酰胺类β晶型成核剂。利用该方法不能制备出本发明不对称取代酰胺类β晶型成核剂。而本发明将对位上一端的基团进行保护后,让另一端的基团先与胺进行反应,再通过分步合成制备得到不对称取代酰胺类β晶型成核剂。
[0057]
进一步地,所述催化剂分别选自1
‑
(3
‑
二甲氨基丙基)
‑3‑
乙基碳二亚胺盐酸盐(edc.hcl)、4
‑
二甲氨基吡啶(dmap)、锡粉、铁粉、镍粉、锌粉、水合肼、二水氯化亚锡中的至少一种;
[0058]
所述有机溶剂选自无水乙醇、n,n
′‑
二甲基甲酰胺(dmf)、二氯甲烷、乙酸乙酯中的
至少一种。
[0059]
需要说明的是,上述催化剂和有机溶剂的选取并不限于此,凡是采用本发明提供的制备方法在选取其他不同的催化剂或者有机溶剂的条件下制备得到具有上述结构的成核剂,均属于本发明的范围。
[0060]
在一些实施方式中,所述a为取代或未取代的c1‑
12
亚烷基、c5‑
12
亚环烷基、c2‑
12
亚烯基、c5‑
12
亚环烯基、亚苯基、2,6
‑
亚萘基、亚联苯基中的一种;
[0061]
r1和r2不同,分别独立地选自取代或未取代的c1‑
12
烷基、c5‑
12
环烷基、c5‑
12
环烯基、苯基、c5‑
12
杂环基中的一种。
[0062]
进一步地,所述杂环基包括含n、o或s原子中的一个或多个。
[0063]
在一些实施方式中,所述a任选被一个或多个以下取代基取代:c1‑
12
烷基、c1‑
12
烷氧基、c1‑
12
烷硫基、c1‑
12
烷基氨基、c1‑
12
烷基磺酰基、c1‑
12
烷基羰基、c1‑
12
烷氧基羰基、醛基、羧基、氰基、氨基、硝基、羟基、巯基、磺基和卤素;
[0064]
所述r1和r2任选被一个或多个以下取代基取代:c1‑
12
烷基、c1‑
12
烷氧基、c1‑
12
烷硫基、c1‑
12
烷基氨基、c1‑
12
烷基磺酰基、c1‑
12
烷基羰基、c1‑
12
烷氧基羰基、醛基、羧基、氰基、氨基、硝基、巯基、磺基和卤素。
[0065]
本发明的成核机理:等规聚丙烯β晶型成核剂表现出的β晶成核能力源于成核剂晶体与等规聚丙烯β晶之间存在链轴和链间两个维度的晶格匹配关系,本发明制备出的成核剂晶体与等规聚丙烯β晶之间晶格匹配的失配率较低,因此表现出高效的β晶成核效率。
[0066]
另外,成核剂的β晶成核效率与中心基团的类型及尺寸、外围基团的类型、尺寸及长度、分子对称性、成核剂的晶体形貌、晶体尺寸及空间分布状态有关,通过调控成核剂的结构与形貌可优化成核剂的β晶成核效率。
[0067]
本发明再一目的提供了上述任一所述的一类高效等规聚丙烯β晶型成核剂在制备β晶型等规聚丙烯中的应用。
[0068]
在一些实施方式中,将等规聚丙烯、抗氧化剂和β晶型成核剂进行熔融共混、造粒,并经过加工得到β晶型等规聚丙烯制品;
[0069]
优选地,所述加工的方式选自注塑、挤出、吹塑、热成型、发泡中的至少一种。
[0070]
进一步地,所述抗氧化剂采用受阻酚类、亚磷酸酯类抗氧剂、硫酯类抗氧剂中的至少一种,用量为等规聚丙烯质量的0.10~0.20wt%;
[0071]
所述等规聚丙烯β晶型成核剂的用量为等规聚丙烯质量的0.03~0.30wt%;
[0072]
优选地,所述等规聚丙烯β晶型成核剂的用量为等规聚丙烯质量的0.05~0.20wt%。
[0073]
采用上述技术方案后,本发明与现有技术相比具有以下有益效果:
[0074]
本发明提供的一类等规聚丙烯β晶型成核剂,即等规聚丙烯不对称取代酰胺类β晶型成核剂可以高效诱导β晶型聚丙烯的生成,用x射线衍射xrd测定,等规聚丙烯β晶型的相对含量达85~99%。用差示扫描量热分析dsc测定,等规聚丙烯β晶型的相对含量达87~99%。
[0075]
本发明提供的一类等规聚丙烯β晶型成核剂,即等规聚丙烯不对称取代酰胺类β晶型成核剂,经其改性后的聚丙烯样品与未改性的聚丙烯样品相比,热变形温度提高了15~19℃,冲击性能和断裂伸长率均提高3~4倍;与经对称取代酰胺类β晶型成核剂(dcht)改性
后的等规聚丙烯样品相比,经本发明制备出的成核剂改性后的聚丙烯样品β晶型的相对含量也提高了5~16%,诱导生成β晶能力更强。热变形温度提高了2~6℃,冲击性能提高了12~39%,断裂伸长率提高了12~37%。
[0076]
本发明提供的一类等规聚丙烯β晶型成核剂,即等规聚丙烯不对称取代酰胺类β晶型成核剂,经其改性后的聚丙烯样品与未改性的聚丙烯样品相比,结晶温度提高了7~12℃;与经对称取代酰胺类β晶型成核剂(dcht)改性后的等规聚丙烯样品相比,结晶温度提高了0.1~2℃,有利于缩短产品的成型周期,提高生产效率。
[0077]
本发明提供的一类等规聚丙烯β晶型成核剂,即等规聚丙烯不对称取代酰胺类β晶型成核剂适用于等规聚丙烯均聚物、共聚物、抗冲共聚聚丙烯、等规聚丙烯与弹性体的共混物、聚丙烯/无机填料共混物和等规聚丙烯/弹性体/无机填料的共混物等体系。
[0078]
本发明提供的一类等规聚丙烯β晶型成核剂,即等规聚丙烯不对称取代酰胺类β晶型成核剂,与聚丙烯相容性好,在聚丙烯熔体中分布均匀,在使用时加工窗口宽,热稳定性好,在聚丙烯常规加工温度200~260℃范围内,以及常规加工压力下,可诱导等规聚丙烯形成含有85~99%的β晶。可广泛应用于pp
‑
r、pp
‑
rct管材、土工格栅、汽车用保险杠、蓄电池槽和热水管料、家电及其他要求高抗冲击性和高热变形性的聚丙烯制品,亦适用于微孔聚丙烯纤维、包装透气膜、干法锂电池隔膜等,拓展了等规聚丙烯的应用领域,极具工业应用前景。
[0079]
下面对本发明的具体实施方式作进一步详细的描述。
具体实施方式
[0080]
为使本发明实施例的目的、技术方案和优点更加清楚,下面对实施例中的技术方案进行清楚、完整地描述,以下实施例用于说明本发明,但不用来限制本发明的范围。
[0081]
实施例1
[0082]
制备的具体步骤如下:
[0083]
(1)向圆底烧瓶中加入1.6g 6
‑
甲氧基
‑6‑
氧代己酸、0.9g环戊胺、和1.2g 4
‑
二甲氨基吡啶(dmap),然后再加入30ml二氯甲烷,常温搅拌条件下分批加入2g 1
‑
(3
‑
二甲氨基丙基)
‑3‑
乙基碳二亚胺盐酸盐(edc.hcl)。常温搅拌18h后,加热回流2~3h,加热温度为60℃。再向反应体系中加入稀盐酸水溶液酸化至ph为7,再利用二氯甲烷进行萃取,收集有机相,通过旋蒸除去有机相中的二氯甲烷后,可得粗产品6
‑
(环戊基氨基)
‑6‑
氧代己酸甲酯。
[0084]
(2)向圆底烧瓶中加入2.3g 6
‑
(环戊基氨基)
‑6‑
氧代己酸甲酯、0.8g氢氧化钠、20ml水和40ml无水乙醇,加热回流5~6h,加热温度为100℃,直至点板检测原料消失。然后通过旋蒸除去乙醇,再向剩余物中加入稀盐酸酸化至ph为7,将析出的固体物质进行抽滤获得粗产品6
‑
(环戊基氨基)
‑6‑
氧代己酸。
[0085]
(3)向圆底烧瓶中加入2.1g干燥后的6
‑
(环戊基氨基)
‑6‑
氧代己酸、1.2g环己胺、1.2g 4
‑
二甲氨基吡啶(dmap),然后再加入70~80ml二氯甲烷。常温搅拌条件下分批加入2g 1
‑
(3
‑
二甲氨基丙基)
‑3‑
乙基碳二亚胺盐酸盐(edc.hcl)。常温搅拌18h后,加热回流3h,加热温度为60℃。通过旋蒸除去二氯甲烷,再向反应体系中加入稀盐酸水溶液酸化至ph为7,
经抽滤获得固体物质,将固体物质用少量二氯甲烷淋洗后,将剩余固体物质再用二甲基亚砜(dmso)进行重结晶,最终制备出1.8g n
‑
环戊基
‑
n
′‑
环己基己二酰胺。产率:61%。
[0086]
实施例2
[0087]
的制备:
[0088]
将实施例1中的环己胺替换成苯胺,具体制备步骤同实施例1,制备出2g n
‑
环戊基
‑
n
′‑
苯基己二酰胺,产率:64%。
[0089]
实施例3
[0090]
的制备:
[0091]
将实施例1中的6
‑
甲氧基
‑6‑
氧己酸替换成4
‑
(甲氧基羰基)环己烷
‑1‑
甲酸,具体制备步骤同实施例1,制备出2.1g n
‑
环戊基
‑
n
′‑
环己基
‑1‑4‑
环己烷二甲酰胺,产率:66%。
[0092]
实施例4
[0093]
的制备:
[0094]
将实施例1中的6
‑
甲氧基
‑6‑
氧己酸替换成4
‑
(甲氧基羰基)环己烷
‑1‑
甲酸,将环己胺替换成苯胺,具体制备步骤同实施例1,制备出1.9g n
‑
环戊基
‑
n
′‑
苯基
‑1‑4‑
环己烷二甲酰胺,产率:60%。
[0095]
实施例5
[0096]
的制备:
[0097]
将实施例1中的6
‑
甲氧基
‑6‑
氧己酸替换成4
‑
(甲氧基羰基)苯甲酸,具体制备步骤同实施例1,制备出1.8g n
‑
环戊基
‑
n
′‑
环己基对苯二甲酰胺,产率:64%。
[0098]
实施例6
[0099]
的制备:
[0100]
将实施例1中的6
‑
甲氧基
‑6‑
氧己酸替换成4
‑
(甲氧基羰基)苯甲酸,将环己胺替换成苯胺,具体制备步骤同实施例1,制备出1.9g n
‑
环戊基
‑
n
′‑
苯基对苯二甲酰胺,产率:61%。
[0101]
实施例7
[0102]
的制备:
[0103]
将实施例1中的6
‑
甲氧基
‑6‑
氧己酸替换成6
‑
(甲氧基羰基)
‑2‑
萘甲酸,具体制备步骤同实施例1,制备出2.5g n
‑
环戊基
‑
n
′‑
环己基
‑2‑6‑
萘二甲酰胺,产率:69%。
[0104]
实施例8
[0105]
的制备:
[0106]
将实施例1中的6
‑
甲氧基
‑6‑
氧己酸替换成6
‑
(甲氧基羰基)
‑2‑
萘甲酸,将环己胺替换成苯胺,具体制备步骤同实施例1,制备出2.3g n
‑
环戊基
‑
n
′‑
苯基
‑2‑6‑
萘二甲酰胺,产率:64%。
[0107]
实施例9
[0108]
的制备:
[0109]
将实施例1中的6
‑
甲氧基
‑6‑
氧己酸替换成(e)
‑6‑
甲氧基
‑6‑
氧代己
‑3‑
烯酸,具体制备步骤同实施例1,制备出2g(e)
‑
n
‑
环戊基
‑
n
′‑
环己基己
‑3‑
烯二酰胺,产率:64%。
[0110]
实施例10
[0111]
的制备:
[0112]
将实施例1中的6
‑
甲氧基
‑6‑
氧己酸替换成4
‑
(甲氧基羰基)环己
‑
2,5
‑
二烯
‑1‑
羧酸,具体制备步骤同实施例1,制备出2g n
‑
环戊基
‑
n
′‑
环己基环己
‑
2,5
‑
二烯
‑
1,4
‑
二甲酰胺,产率:63%。
[0113]
实施例11
[0114]
的制备:
[0115]
将实施例1中的6
‑
甲氧基
‑6‑
氧己酸替换成4
′‑
(甲氧基羰基)
‑
[1,1
′‑
联苯]
‑4‑
羧酸,具体制备步骤同实施例1,制备出2.3g n
‑
环戊基
‑
n
′‑
环己基
‑
[1,1
′‑
联苯]
‑
4,4
′‑
二甲酰胺,产率:64%。
[0116]
实施例12
[0117]
的制备:
[0118]
将实施例1中的6
‑
甲氧基
‑6‑
氧己酸替换成4
‑
(甲氧基羰基)苯甲酸,将环戊胺替换成戊胺,具体制备步骤同实施例1,制备出2g n
‑
戊基
‑
n
′‑
环己基
‑
对苯二甲酰胺,产率:64%。
[0119]
实施例13
[0120]
的制备:
[0121]
将实施例1中的6
‑
甲氧基
‑6‑
氧己酸替换成4
‑
(甲氧基羰基)苯甲酸,将环戊胺替换成环己
‑
2,5
‑
二烯
‑1‑
胺,具体制备步骤同实施例1,制备出2.1g n
‑
(环己
‑
2,5
‑
二烯
‑1‑
基)
‑
n
′‑
环己基
‑
对苯二甲酰胺,产率:67%。
[0122]
实施例14
[0123]
的制备:
[0124]
将实施例1中的6
‑
甲氧基
‑6‑
氧己酸替换成4
‑
(甲氧基羰基)苯甲酸,将环戊胺替换成4
‑
氨基吡啶,具体制备步骤同实施例1,制备出2g n
‑
环己基
‑
n
′‑
(吡啶
‑4‑
基)对苯二甲酰胺对苯二甲酰胺,产率:64%。
[0125]
实施例15
[0126]
的制备:
[0127]
将实施例1中的6
‑
甲氧基
‑6‑
氧己酸替换成4
‑
(甲氧基羰基)
‑2‑
甲基苯甲酸,具体制备步骤同实施例1,制备出2.1g n
‑
环戊基
‑
n
′‑
环己基
‑2‑
甲基对苯二甲酰胺,产率:64%。
[0128]
实施例16
[0129]
的制备:
[0130]
将实施例1中的6
‑
甲氧基
‑6‑
氧己酸替换成4
‑
(甲氧基羰基)苯甲酸,将环己胺替换成4
‑
甲基环己胺,具体制备步骤同实施例1,制备出2.1g n
‑
环戊基
‑
n
′‑
(4
‑
甲基环己基)对苯二甲酰胺,产率:64%。
[0131]
实施例17
[0132]
制备的具体步骤如下:
[0133]
(1)向圆底烧瓶中加入1.4g对硝基苯胺、1.3g环己基羧酸、1.2g 4
‑
二甲氨基吡啶(dmap),然后再加入30ml二氯甲烷,常温搅拌条件下分批加入2g 1
‑
(3
‑
二甲氨基丙基)
‑3‑
乙基碳二亚胺盐酸盐(edc.hcl)。常温搅拌18h后,加热回流2~3h,加热温度为60℃。再向反应体系中加入稀盐酸水溶液酸化至ph为7,再利用二氯甲烷进行萃取,收集有机相,通过旋蒸除去有机相中的二氯甲烷后,可得粗产品n
‑
(4
‑
硝基苯基)环己烷甲酰胺;
[0134]
(2)向圆底烧瓶中加入2.5g n
‑
(4
‑
硝基苯基)环己烷甲酰胺,1g锡粉,常温搅拌条件下分批加入50ml浓盐酸溶液。加热回流2h,加热温度为100℃。冷却至室温,再向反应体系中加入氢氧化钠溶液使反应体系呈碱性。旋蒸直至出澄清液为止,将馏出液放入分液漏斗中,分出粗产品n
‑
(4
‑
氨基苯基)环己烷甲酰胺。向水层加入氯化钠,用乙醚分两次萃取,合
并n
‑
(4
‑
氨基苯基)环己烷甲酰胺和乙醚萃取液,用naoh颗粒干燥,制备出n
‑
(4
‑
氨基苯基)环己烷甲酰胺;
[0135]
(3)向圆底烧瓶中加入2.2g干燥后的n
‑
(4
‑
氨基苯基)环己烷甲酰胺、1.1g环戊基羧酸、1.2g 4
‑
二甲氨基吡啶(dmap),然后再加入70~80ml二氯甲烷。常温搅拌条件下分批加入2g 1
‑
(3
‑
二甲氨基丙基)
‑3‑
乙基碳二亚胺盐酸盐(edc.hcl)。常温搅拌18h后,加热回流3h,加热温度为60℃。通过旋蒸除去二氯甲烷,再向反应体系中加入稀盐酸水溶液酸化至ph为7,经抽滤获得固体物质,将固体物质用少量二氯甲烷淋洗后,将剩余固体物质再用二甲基亚砜(dmso)进行重结晶,最终制备出2g n
‑
(4
‑
(环戊烷甲酰胺基)苯基)环己基甲酰胺。产率:65%
[0136]
实施例18
[0137]
制备的具体步骤如下:
[0138]
(1)向圆底烧瓶中加入1.0g环己胺、1.4g对氨基苯甲酸、1.2g 4
‑
二甲氨基吡啶(dmap),然后再加入30ml二氯甲烷,常温搅拌条件下分批加入2g 1
‑
(3
‑
二甲氨基丙基)
‑3‑
乙基碳二亚胺盐酸盐(edc.hcl)。常温搅拌18h后,加热回流2~3h,加热温度为60℃。再向反应体系中加入稀盐酸水溶液酸化至ph为7,再利用二氯甲烷进行萃取,收集有机相,通过旋蒸除去有机相中的二氯甲烷后,可得粗产品4
‑
氨基
‑
n
‑
环己基苯甲酰胺;
[0139]
(2)向圆底烧瓶中加入2.2g干燥后的4
‑
氨基
‑
n
‑
环己基苯甲酰胺、1.1g环戊基羧酸、1.2g 4
‑
二甲氨基吡啶(dmap),然后再加入70~80ml二氯甲烷。常温搅拌条件下分批加入2g 1
‑
(3
‑
二甲氨基丙基)
‑3‑
乙基碳二亚胺盐酸盐(edc.hcl)。常温搅拌18h后,加热回流3h,加热温度为60℃。通过旋蒸除去二氯甲烷,再向反应体系中加入稀盐酸水溶液酸化至ph为7,经抽滤获得固体物质,将固体物质用少量二氯甲烷淋洗后,将剩余固体物质再用二甲基亚砜(dmso)进行重结晶,最终制备出2.1g n
‑
环己基
‑4‑
(环戊烷甲酰胺基)苯甲酰胺。产率:67%
[0140]
以下通过实验例对本发明成核剂和对比例成核剂的β晶成核效率作进一步说明。但这里提供的只是本发明的优选实施方式,本发明的范围并不受此限制。
[0141]
以下实验例所使用的等规聚丙烯(牌号为s1003,熔体流动速率为2.5g/10min)由中国石化北京燕山石油化工有限公司生产。
[0142]
实验例1
[0143]
样品1#的制备:将等规聚丙烯粒料,0.1wt%抗氧剂(irganox b 215)分别和0.05wt%、0.10wt%、0.20wt%表1中序号1的成核剂在高速混合机中搅拌10分钟使其混合均匀得到预混料,使用双螺杆挤出机将预混料混炼、挤出、造粒,然后再注塑制得样品1#。其中,挤出温度为180
–
210℃,注塑成型机各段温度220
–
230℃。
[0144]
实验例2
‑
32
[0145]
样品2#
‑
样品32#的制备:与实验例1的区别在于,将序号1的成核剂依次替换成表1
‑
表3中序号2、7、8、13、14、19、20、25、29、33、34、35、36、37、38、45、46、51、52、63、67、83、84、85、86、95、96、97、98、119、123的成核剂,其他实验条件完全相同。
[0146]
实验例33
[0147]
对比样1#的制备:将等规聚丙烯粒料和0.1wt%抗氧剂(irganox b 215)在高速混合机中搅拌10分钟使其混合均匀后,使用双螺杆挤出机将预混料混炼、挤出、造粒,然后再注塑制得制得对比样1#。挤出温度为180
–
210℃,注塑成型机各段温度220
–
230℃。
[0148]
实验例34
[0149]
对比样2#的制备:将等规聚丙烯粒料,0.1wt%抗氧剂(irganox b 215)分别和0.05wt%、0.10wt%、0.20wt%对称取代酰胺类β晶型成核剂dcht(山西省化工研究所(有限公司)产品)在高速混合机中搅拌10分钟使其混合均匀后,使用双螺杆挤出机将预混料混炼、挤出、造粒,然后再注塑制得制得对比样2#。挤出温度为180
–
210℃,注塑成型机各段温度220
–
230℃。
[0150]
实验例35
‑
37
[0151]
对比样3#
‑
对比样5#的制备:与实验例34的区别在于,将1,4
‑
二环己基对苯二甲酰胺(dcht)成核剂依次替换成稀土类β晶型成核剂wbg(广东炜林纳新材料科技股份有限公司产品)、有机金属盐类聚丙烯β晶型成核剂nab
‑
83(广州呈和有限公司产品)、ca
‑
pim(氢氧化钙与庚二酸酸碱中和反应制得),其他实验条件完全相同,制备出对比样3#
‑
对比样5#。
[0152]
将如上实验例1
‑
37制备得到的等规聚丙烯样品中β晶型的相对含量测定:
[0153]
本发明采用ta公司q200型热分析仪测定等规聚丙烯样品中β晶型的相对含量。测试前用金属铟进行温度校正,将样品由室温快速升温至230℃,等温5min消除热历史,以10℃/min降温至100℃等温1min,再以10℃/min升温至200℃,记录结晶曲线和二次熔融曲线。等规聚丙烯β晶型的相对含量计算公式如下:
[0154][0155]
其中,δh
β
为β晶型等规聚丙烯的熔融焓;δh
α
为α晶型等规聚丙烯的熔融焓。
[0156]
本发明亦采用xrd测定等规聚丙烯样品中β晶型的相对含量。xrd实验在法国xenocs公司的的xeuss 2.0系统上进行。cukα源波长为散射花样通过二维面探测器(pilatus,300k dectris)收集,分辨率为487
×
619像素,像素尺寸为172μm
×
172μm,样品到检测器的距离为127mm,曝光时间为10min。等规聚丙烯β晶型的相对含量计算公式如下:
[0157][0158]
其中,i
β
(100)为β晶型(110)晶面对应衍射峰的峰强;i
α
(110),i
α
(040),i
α
(130)分别为α晶(110)、(040)和(130)晶面对应衍射峰的峰强。
[0159]
实验例1
‑
37各样品的结晶温度及等规聚丙烯β晶型的相对含量的测试结果见表4。
[0160]
表4
[0161]
[0162]
[0163]
[0164][0165]
将如上实验例1
‑
37制备得到的聚丙烯样品进行相关性能测试,测试的相关性能如下:
[0166]
(1)缺口冲击强度(kj/m2),测试方法:参照悬臂梁缺口冲击强度测试执行标准gb/t1843
‑
1996;
[0167][0168]
(2)断裂伸长率(%),测试方法:参照gb/t 1040
‑
92;
[0169]
(3)热变形温度(℃),测试方法:参照gb/t 1634
‑
2004;
[0170]
等规聚丙烯的相关性能测试按gb 2918
‑
1998(塑料试样状态调节和试验的标准环境)规定,在(23
±
2)℃,相对湿度为(50
±
5)%的条件下进行,试样状态调节时间48h。
[0171]
实验例1
‑
34各样品的缺口冲击强度、断裂伸长率及热变形温度的测试结果见表5。
[0172]
表5
[0173]
[0174]
[0175]
[0176][0177]
通过表4和表5的数据可以看出,采用本技术提供的成核剂能够制备出具有极高β晶型含量的等规聚丙烯,在成核剂的添加量为0.05~0.20wt%时,用差示扫描热分析dsc测定,等规聚丙烯β晶型的相对含量达87~99%。用x射线衍射xrd测定,等规聚丙烯β晶型的相对含量达85~99%。这是由于本发明制备出的成核剂晶体与等规聚丙烯β晶之间的失配率较低,并通过调控成核剂形貌优化了成核剂的β晶成核效率,因此表现出高效的β晶成核效率。
[0178]
通过表4和表5的数据可以看出,经本发明制备出的成核剂改性后的等规聚丙烯样品与未改性的等规聚丙烯样品相比,其结晶温度提高了7~12℃,冲击性能和断裂伸长率均提高3~4倍,热变形温度提高了15~19℃。与经对称取代酰胺类β晶型成核剂(dcht)改性后的等规聚丙烯样品相比,其结晶温度提高了0.1~2℃,用dsc和xrd测定的本发明等规聚丙烯β晶型的相对含量提高了5~16%。冲击性能提高了12~39%,断裂伸长率提高了12~37%,热变形温度最高提高了2~6℃。这是由于本发明制备得到的β晶成核剂在聚丙烯基体中分散良好,诱导形成的β晶含量极高,从而提高了聚丙烯样品的抗冲击性能和断裂伸长率和热变形温度。
[0179]
以上所述仅是本发明的较佳实施例而已,并非对本发明作任何形式上的限制,虽然本发明已以较佳实施例揭露如上,然而并非用以限定本发明,任何熟悉本专利的技术人员在不脱离本发明技术方案范围内,当可利用上述提示的技术内容作出些许更动或修饰为等同变化的等效实施例,但凡是未脱离本发明技术方案的内容,依据本发明的技术实质对以上实施例所作的任何简单修改、等同变化与修饰,均仍属于本发明方案的范围内。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1