用于嵌合抗原受体分子的调控表达的慢病毒载体
1.本发明属于重组疫苗技术的领域并涉及慢病毒载体的改进,其可用作治疗性和预防性疫苗。该载体提供优于其他载体的改善的免疫应答诱导。
背景技术:
2.重组疫苗已随重组dna技术的进步而发展,允许修饰病毒基因组以产生修饰的病毒。通过这种方式,已能够将遗传序列引入非致病性病毒,从而它们编码需要在感染或转导后于靶细胞内得以表达的免疫原性蛋白质,以在其宿主内发展特定免疫应答。
3.这类载体构成了疫苗技术的主要优势(kutzler等,nat rev genet,9(10):776
‑
788,2008)。具体而言,它们具有优于传统疫苗的优势:避免活(减毒的)病毒和消除灭活疫苗制造的相关风险。
4.在二十世纪八十年代早期由mann等引入使用改性的逆转录病毒(逆转录病毒载体)的基因递送(cell,33(1):153
‑
9,1983)。最常用的致瘤性逆转录病毒基于莫洛尼(moloney)鼠白血病病毒(mlv)。它们具有简单的基因组,从中产生多蛋白gag、pol和env并且要求为反式以用于病毒复制(breckpot等,2007,gene ther,14(11):847
‑
62;he等,2007,expert rev vaccines,6(6):913
‑
24)。一般需要的顺式序列是长末端重复序列(ltr)及其周边序列:用于整合的反向重复序列(ir或att位点)、包装序列ψ、转运rna结合位点(引物结合位点,pbs),和涉及逆转录的一些其它序列(正链起始必需的ltr内的重复r以及多嘌呤序列(ppt))。为了生成复制缺陷型逆转录病毒载体,通常使gag、pol和env基因全部缺失,并且用表达盒替代。
5.源自慢病毒基因组的逆转录病毒载体(即,慢病毒载体)已涌现为用于基因治疗和免疫治疗目的的有希望的工具,因为它们显示优于其它病毒系统的若干优势。具体地,慢病毒本身没有毒性,并且与其它逆转录病毒不同,慢病毒能够转导非分裂细胞,尤其是树突状细胞(he等,2007,expert rev vaccines,6(6):913
‑
24),使得通过内源性途径呈递抗原。
6.慢病毒通过遗传组成相似性、复制的分子机制和与其宿主的生物相互作用连接。它们最常称为缓慢疾病综合征的试剂,该综合征在延长的亚临床感染之后开始并缓慢发展;因此,它们被称作“慢”病毒(narayan等,1989,j gen virol,70(7):1617
‑
39)。其基本构成与所用逆转录病毒相同但由于附属基因(例如vif、vpr、vpu、nef、tat和rev)的存在而更为复杂,所述附属基因在慢病毒的体内复制中起关键作用。
7.慢病毒代表逆转录病毒科(retroviridae)的一个慢病毒(slow virus)属,其包括人免疫缺陷病毒(hiv)、猿猴免疫缺陷病毒(siv)、马传染性脑炎病毒(eiav)、山羊关节炎脑炎病毒(caev)、牛免疫缺陷病毒(biv)和猫免疫缺陷病毒(fiv)。慢病毒可以无限地持续存在于其宿主内,并且在一生的感染期间以不同速度连续复制。病毒在其宿主内的持续复制取决于其避开宿主防御的能力。
8.重组整合型慢病毒载体的设计基于慢病毒的顺式与反式作用序列的分离。非分裂细胞中的有效转导需要慢病毒基因组中存在两种顺式作用序列:中心多嘌呤序列(cppt)和
中心终止序列(cts)。它们导致形成三链dna结构,称为中心dna“瓣(flap)”,该结构使基因进入非分裂细胞(包括树突细胞(dc))核内的效率最大化(zennou等,2000,cell,101(2)173
‑
85;arhel等,2007,embo j,26(12):3025
‑
37)。
9.树突细胞对于抗原递呈而言具有重要作用,因为其构成了以递呈抗原和起始免疫应答为主要功能的抗原递呈细胞(apc)的主要类型。
10.为产生免疫应答,须由细胞将抗原蛋白加工成肽,所述肽通过主要组织相容性复合物蛋白(mhc)在细胞表面显示。循环的apc在引流淋巴结内将肽
‑
mhc复合物递呈至t细胞,其在此处与t细胞受体相互作用并联合共刺激信号一起激活t细胞。
11.多项研究已显示,使用慢病毒载体的接种导致dc进行抗原呈递和抗原特异性细胞毒性t淋巴细胞(ctl;cd8
+
t细胞)的强激活。因此,在过去的10年中,慢病毒载体已经工程改造用于基因转化和免疫疗法应用。
12.载体通常含有含增强子的强组成型启动子,如cmv启动子。michelini等,vaccine 27(34):4622
‑
29(2009);karwacz等,j.virol.83(7):30943103(2009);negri等,molecular therapy 15(9):1716
‑
23(2007);和buffa等,j.general virology 87:1625
‑
1634(2006)。
13.已通过移除ltr u3序列,产生完全不含原先在ltr中存在的病毒启动子和增强子序列的“自动失活”载体,从而改善了慢病毒载体的安全性。
14.可通过在不同的dna质粒瞬时转染细胞,例如hek 293t人培养细胞后由重组技术产生含有慢病毒载体的慢病毒颗粒:
15.(i)包装质粒,其表达至少gag、pol、rev、tat,并在一些情况下表达转移构建体的包装所必需的结构和酶蛋白;
16.(ii)原病毒转移质粒,其含有包装、逆转录和整合所必需的表达盒和hiv顺式作用因子;以及
17.(iii)编码包膜的质粒,在多数情况下是水泡性口炎病毒(vsv.g)的糖蛋白,该蛋白允许形成能够靶向多种细胞(尤其是主要组织相容性(mhc)抗原递呈细胞(apc),包括dc的混合颗粒(假型)。
18.该方法能够使转染的细胞瞬时生产慢病毒颗粒载体。然而,也可通过向细胞基因组中稳定地插入包装基因、原病毒编码dna和包膜基因来连续产生慢病毒颗粒载体。这使得能够由细胞连续产生慢病毒颗粒载体而不需要瞬时转染。当然,也可使用这些方法的组合,其中将一些dna/质粒整合至细胞基因组而其他的通过瞬时转染提供。
19.非整合型慢病毒载体已被设计用于降低与插入诱变事件相联系的潜在癌发生的风险,特别用于疫苗接种目的。非整合型慢病毒载体的示例提供于coutant等,plos one 7(11):e48644(2102),karwacz等,j.virol.83(7):3094
‑
3103(2009),negri等,molecular therapy 15(9):1716
‑
1723(2007);hu等,vaccine 28:6675
‑
6683(2010)。因此,已有报告称与整合型系统相比,非整合型慢病毒载体系统可降低插入型诱变的潜在风险。hu等,vaccine 28:6675
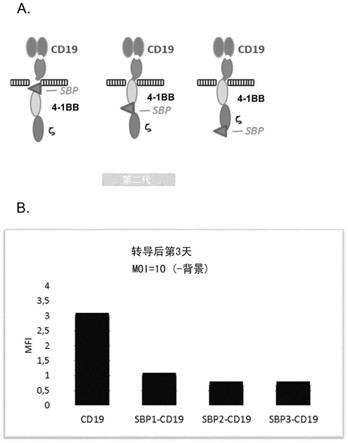
‑
6683(2010)。
20.此外,自动失活的慢病毒载体中病毒启动子和增强子序列的3
′
ltr的u3区域的缺失限制了内源性启动子激活的可能性。这些对于安全性的关注直接体现了1998
‑
1999年进行的scid
‑
x1基因治疗试验所获得的经验,该试验使用基于莫罗尼病毒的逆转录病毒载体针对患有罕见类型的x连锁(scid
‑
x1基因)严重免疫缺陷疾病的儿童进行(cavazzana
‑
calvo等,2000,science.,288(5466):669
‑
72)。在该试验中,9名儿童中的4名由于莫罗尼衍生的慢病毒载体在非常靠近人lm02原癌基因处整合而发展出白血病(hacein
‑
bey
‑
abina等,2008,j.clin.invest.,118(9):3132
‑
3142)。这表明,恶性肿瘤是病毒u3启动子/增强子接近lm02原癌基因的结果。结果,安全性是向人给予慢病毒载体的主要问题。
21.增强子是顺式作用序列,其可相隔一定距离作为转录激活因子起作用。它们已经在病毒衍生的载体中广泛采用,因为它们似乎对于在多种细胞类型,尤其是dc中获得转基因强表达而言最高效(chinnasamy等,2000,hum gene ther 11(13):1901
‑
9;rouas等,2008,cancer gene ther 9(9):715
‑
24;kimura等,2007,mol ther 15(7):1390
‑
9;gruh等,2008,j gene med 10(1)21
‑
32)。然而,考虑到插入诱变的安全问题,应将这类转录增强子序列从慢病毒载体构建体中删除以破坏增强子邻近效应所产生的插入诱变风险。迄今为止,该增强子邻近效应是最常见的插入突变机制并且是基因转移后人或动物肿瘤发生事件的病例中描述的唯一效应。
22.因此,需要研发不包括病毒增强子但仍然允许转基因编码的免疫原性肽足量表达的逆转录病毒,特别是慢病毒载体,如果可能,表达量与使用cmv启动子时观察到的相当。
23.最近的研究报道了使用主要组织相容性复合物ii型基因(mhc ii型)(kimura等,2007,molther 15(7):1390
‑
9)和dectin
‑
2基因(lopes等,2008,j virol 82(1):86
‑
95)来源的dc特异性启动子代替病毒启动子。lopes等使用的dectin
‑
2基因启动子包含推定的增强子和腺病毒保守序列(腺病毒启动子中的反向末端重复)(bonkabara等,2001,j.immunology,167:6893
‑
6900)。kimura等使用的mhc ii型基因启动子不含任何已知增强子。
24.然而,当在静脉内给予时,在没有增强子的情况下,发现mhc ii型启动子无法在dc中提供足够的转基因表达。具体而言,与使用cmv启动子/增强子所观察到的免疫应答相反,包含mhc ii型启动子的慢病毒载体未能在免疫活性c57bl/6小鼠中引起免疫反应。虽然在小鼠中注射后观察到了整合和持续的转基因表达,但通过mhc ii型启动子转录的慢病毒载体无法刺激抗原特异性cd8+细胞毒性t淋巴细胞应答,甚至在疫苗接种加强后也是如此。因此,这些研究的作者的结论是使用mhc ii型启动子仅对于在基因替换疗法中寻求持续表达的应用而言有兴趣,但在免疫疗法中没有兴趣。应注意,mhc ii型启动子在大多数细胞类型中弱表达。
25.因此,对于通过iv注射诱导针对抗原的免疫应答,mhc ii型启动子不是适当的慢病毒载体启动子。此外,dectin
‑
2启动子在大多数细胞类型中弱表达并且似乎含有增强子。因此,由于安全原因,dectin
‑
2启动子不是良好的慢病毒载体启动子。
26.优选地,在免疫治疗中,慢病毒载体提供了有效的转基因表达,其引发所需的特异性免疫应答。这需要apc(如树突细胞)中的表达处于高水平。
27.还优选通过免疫应答除去慢病毒载体所转导的细胞以提供更高水平的安全性。即,针对转基因产生的免疫应答可在宿主中引发足以除去慢病毒载体转导细胞的免疫应答。消除经转导的细胞消除了慢病毒载体在宿主中的持续存在,以及该载体的可能二级作用。为了消除经转导的细胞,在非树突细胞中需要一定水平的表达以由免疫应答进行消除。因此,需要适当地表达抗原。
28.同时,启动子应通过原初和记忆t细胞的激活中涉及的关键细胞(即树突细胞)使
免疫刺激最大化并应使导致恶性肿瘤的干细胞中的插入诱变和基因毒性的风险最小化。因此,启动子应在树突和其他细胞中具有足够高的活性,但不含增强子。基于这些标准,由于强增强子的存在,病毒启动子(如cmv启动子)不是理想的。这些标准归纳如下:
29.在抗原递呈细胞(包括树突细胞)中高表达以诱导最大的免疫应答;
30.在其他转导的细胞类型中以足够的水平表达,该表达水平足以供于通过诱导的免疫应答进行消除;以及
31.缺少增强子元件以避免插入效应。
32.嵌合抗原受体(car)是针对抗原的重组受体。sadelain等,cancer discov.2013年4月;3(4):388
‑
398。它们是一般靶向天然细胞表面抗原的重组受体。同上。car涉及不需要识别hla表达或肽加工的分子。同上。car已经显示自身具有t细胞
‑
增殖和活化潜力,包括在cd3
‑
ζ或fc受体γ和cd8、cd4、cd25或cd16之间的嵌合分子。car的抗原结合部分一般是衍生自抗体的scfv,选自文库的fab,或天然受体配体。han eq等,j hematol oncol.6:47,2013。
33.car已经针对许多不同细胞表面分子生成,包括cd19、her2、gd2、psma,并且许多正处于临床试验中。davila等,oncoimmunology 1:9,1577
‑
1583;2012。car的最有希望的临床结果已经出现在用靶向cd19的自体同源car
‑
修饰的t细胞治疗的患者中。maus mv等,123(17):2625
‑
35,2014。
34.已近研究了表达car的t细胞的体内消除。已经用慢病毒载体遗传修饰人t细胞以表达含有自杀基因的cd20
‑
car,其依赖于胱冬酶9的诱导型活化。budde等,plos one 8(12):e82742(2013)。这种慢病毒载体使用人ef1α启动子来获得t细胞中的表达。自杀融合物的活化导致在体外和体内同时高效去除转导的t细胞。同上。
35.如果能够快速关闭细胞表面上的car表达以防止不良事件并且能够在其已关闭之后再激活系统则会更好。因此,本领存在对用于人类治疗改善的方法和载体的需求。本发明满足本领域中的这些需要。
技术实现要素:
36.本发明包括编码car的核酸分子和载体以及制备和使用该核酸分子和载体的方法。在一个实施方式中,本发明包括嵌合抗原受体,其包含结合结构域;跨膜结构域;hook
‑
结合结构域,优选包含链霉亲和素
‑
结合肽;和活化结构域,其包含seq id no:3、seq id no:4、seq id no:5、seq id no:6、seq id no:7、或seq id no:8、seq id no:33、或seq id no:34的至少100个氨基酸的t细胞活化片段。
37.在一个实施方式中,本发明包括表达嵌合抗原受体的慢病毒载体,该嵌合抗原受体包含结合结构域;跨膜结构域;hook
‑
结合结构域,包含链霉亲和素
‑
结合肽;和活化结构域,其包含seq id no:3、seq id no:4、seq id no:5、seq id no:6、seq id no:7、或seq id no:8、seq id no:33、或seq id no:34的至少100个氨基酸的t细胞活化片段。
38.在一个实施方式中,本发明包括编码嵌合抗原受体的核酸载体,该嵌合抗原受体包含结合结构域;跨膜结构域;hook
‑
结合结构域,包含链霉亲和素
‑
结合肽;和活化结构域,其包含seq id no:3、seq id no:4、seq id no:5、seq id no:6、seq id no:7、或seq id no:8、seq id no:33、或seq id no:34的至少100个氨基酸的t细胞活化片段。
39.在一个实施方式中,本发明包括编码嵌合抗原受体的慢病毒载体颗粒,该嵌合抗
原受体包含结合结构域;跨膜结构域;hook
‑
结合结构域,包含链霉亲和素
‑
结合肽;和活化结构域,其包含seq id no:3、seq id no:4、seq id no:5、seq id no:6、seq id no:7、或seq id no:8、seq id no:33、或seq id no:34的至少100个氨基酸的t细胞活化片段。
40.在一个实施方式中,嵌合抗原受体或慢病毒载体或核酸载体或慢病毒载体颗粒包含单链fv抗体或单结构域抗体(即,纳米抗体)。
41.在一个实施方式中,载体编码包含seq id no:31或seq id no:32的氨基酸序列的hook。
42.在一个实施方式中,载体包含β2
‑
微球蛋白、ubi、ef1α、mhci、或mhcii启动子。在一个实施方式中,该载体是dna。
43.在一个实施方式中,慢病毒载体颗粒包含水泡性口炎病毒糖蛋白。在一个实施方式中,慢病毒载体颗粒包含hiv
‑
1亚型d gag和pol蛋白。
44.在一个实施方式中,本发明包括包含本发明的载体的分离的细胞。
45.本发明包括本发明的任意嵌合抗原受体或慢病毒载体或核酸载体或慢病毒载体颗粒诱导人的免疫应答的用途。
46.本发明包括诱导人的免疫应答的方法,包括向人给予由编码嵌合抗原受体的慢病毒载体颗粒转导的细胞或所述慢病毒载体颗粒,并且任选地,随后向该人给予生物素。
47.在一些实施方式中,嵌合抗原受体或慢病毒载体或核酸载体包括seqid no:45、seq id no:47、或seq id no:49中任一个的核苷酸序列。在一些实施方式中,嵌合抗原受体包括seq id no:46、seq id no:48、或seq id no:50中任一个的氨基酸序列。
48.在一些实施方式中,慢病毒载体或核酸载体还编码seq id no:42的氨基酸序列或包含seq id no:43和/或seq id no:44的核酸序列。
49.附图的简要说明
50.图1显示了来自含所示启动子的慢病毒载体在hek 293t和bdcm细胞中的gfp表达。
51.图2显示了使用在biogps.org上的数据分析的各种启动子的细胞特异性表达。表达水平对骨骼肌中的表达(=1.0)进行标准化。
52.图3a
‑
c显示了来自具有人泛素、mhc i型、mhc ii型、和β2微球蛋白(β2m)启动子的各种慢病毒载体的t细胞中car的表达。
53.图4显示了编码与hook
‑
链霉亲和素融合的密码子优化的vsv
‑
g蛋白的假型载体的效价,其具有+/
‑
生物素和hook
‑
链霉亲和素编码载体的剂量梯度。
54.图5a
‑
b显示了car
‑
rush的结构和表达。a)设计的三种car
‑
rush构建方案car
‑
rush是显示4
‑
1bb和cd3ξ的胞内结构域的第二代car。car
‑
rush的特异之处在于在sbp(链霉亲和素结合蛋白)区存在下允许与偶联至链霉亲和素的hook蛋白相互作用,并因此在内质网中停留。sbp1、sbp2和sbp3之间的差异包括sbp区域的位置,其已分别位于跨膜结构域和4
‑
1bb之间、或4
‑
1bb和cd3ζ之间、或c
‑
末端处。b)由facs分析的car
‑
阳性t淋巴细胞的显示中值荧光强度的mfi。细胞在10的moi下转导并在转导后第3天分析。在直方图中报道的mfi值通过从未转导的细胞减去对应于背景的mfi来标准化。所有car
‑
阳性t细胞显示与第二代car
‑
cd19(阳性对照)相当的mfi,该car
‑
cd19显示与其他测试的构建体相比高3倍的mfi。全部三种car
‑
rush(sbp1、sbp2和sbp3)蛋白显示出非常相似的mfi并且因此在细胞表面上以相似水平表达。
55.图6a
‑
b显示了在与编码hook
‑
链霉亲和素的慢病毒载体共转导和生物素处理之后car
‑
rush构建体的表达和性能。由facs分析的car
‑
阳性t淋巴细胞的百分比(a)和显示中值荧光强度的mfi(b)。细胞用编码hook
‑
链霉亲和素的慢病毒载体(moi 5)和编码car
‑
sbp的慢病毒载体(moi 5)共转导,对应于10的最终总moi。在共转导时添加生物素并且在转导后第3天分析细胞。通过从减去对应于背景的来自未转导的细胞的值获得直方图中报道的百分比和mfi值。car
‑
rush构建体在向培养基中添加生物素之后特异性地以低百分比表达,表明在共转导的情况中,car
‑
rush被hook蛋白高效保留并且其能够仅在生物素介导的诱导之后在细胞膜上呈递。
56.图7a
‑
b显示了car
‑
rush双顺反子构建体的表达和性能。由facs分析的car
‑
阳性t淋巴细胞的显示中值荧光强度的mfi和百分比。细胞在10(a)或30(b)的moi下转导并在转导后第3天分析。在转导时添加生物素。通过从减去对应于背景的来自未转导的细胞的值来获得直方图中报道的百分比和mfi值。sbp1
‑
cd19和sbp3
‑
cd19 car
‑
rush构建体仅在培养基中添加生物素之后在细胞表面上显示表达表明,在没有生物素的情况下,car
‑
rush被hook
‑
链霉亲和素蛋白特异性保留,并且在递送生物素之后,rush调控系统诱导他们向质膜的运输以及它们在质膜上的表达。在培养基中添加生物素之后,sbp1
‑
cd19和sbp3
‑
cd19 car
‑
rush构建体在细胞表面上显示表达。在高moi(moi=30)并且甚至在没有生物素的情况下,sbp1
‑
cd19显示轻微的细胞表面表达。鉴于这些结果,car
‑
rush被认为在没有生物素的情况下很大程度上被hook链霉亲和素蛋白保留。此外,如预期的那样,生物素递送诱导它们向质膜的运输以及它们在质膜上的表达。
57.图8a
‑
b显示car
‑
rush系统切换评价(关/开)。在第0天用编码car
‑
rush系统的慢病毒转导纯化的t细胞并且在同时加入或不加入生物素。在第3天,通过流式细胞术分析t细胞中t细胞表面上的car表达。由facs分析的来自一个代表性供体的car
‑
阳性t淋巴细胞的百分比(a)和显示中值荧光强度的mfi(b)。在10或20的moi下转导car
‑
rush构建体,同时添加生物素并且分析t细胞在转导后第3天的细胞表面上的car表达。该实验为了研究car
‑
rush构建体在生物素递送之后的诱导动力学和在没有生物素诱导剂存在下car
‑
rush保留的效率。如上图所示,检测到低百分比的car
‑
阳性t细胞,并且视car
‑
rush构建体而定,效果依赖于或独立于生物素的添加。根据分析的car
‑
rush构建体,检测到低百分比的car
‑
阳性t细胞。针对car
‑
sbp1和car
‑
sbp2检测生物素诱导细胞表面处car
‑
rush表达的能力。car
‑
sbp3在生物素存在或缺失下都显示轻微car表达。
58.图9a
‑
d显示car
‑
rush系统切换评价(关/开/关)。在第0天用编码car
‑
rush系统的慢病毒转导纯化的t细胞并且在同时加入或不加入生物素。在第3天,通过流式细胞术在一组孔中分析在生物素存在或缺失下的car表达。其他组的孔经洗涤并放回含或不含生物素的培养中,以监测car
‑
阳性细胞关闭car的表达的能力。然后在第7天分析t细胞的t细胞表面上的car表达。由facs分析并衍生自健康供体32的car
‑
阳性t淋巴细胞的百分比(a)和显示中值荧光强度的mfi(b)。car
‑
rush构建体在10的moi下转导并在第3天再添加或不添加生物素之后在转导后第7天分析细胞表面上的car表达。如a)中所示,检测到低百分比的car
‑
阳性t细胞。针对car
‑
sbp2和car
‑
sbp3,生物素诱导细胞表面上的car
‑
rush表达,并且尤其是针对car
‑
sbp3,在第3天洗涤和生物素再添加之后仍重新诱导car表达。由facs分析并衍生自健康供体31的car
‑
阳性t淋巴细胞的百分比(c)和显示中值荧光强度的mfi(d)。car
‑
rush构建体在20的moi下转导并在第3天再添加或不添加生物素之后在转导后第7天分析细胞表面上的car表达。全部三种测试的car
‑
rush都显示低百分比的car
‑
阳性细胞。另外,除了在生物素存在或缺失下都显示car表达的car
‑
sbp1以外,car
‑
rush仅在第0天和/或第7天添加生物素的条件中表达。
59.图10a
‑
c显示了car
‑
cd19第二代及其组分的氨基酸和核苷酸序列。a)核苷酸序列(seq id no:39),b)氨基酸序列(seq id no:51),和c)关键处表示信号序列、cd19 scfv、cd8铰链、跨膜结构域、4
‑
1bb、和cd3z序列。
60.图11显示了双顺反子hook链霉亲和素
‑
ires
‑
car
‑
sbp
‑
cd19第2代构建体的结构。
61.发明详述
62.生成编码嵌合抗原受体(car)的多种慢病毒构建体,用于在t细胞中表达car。还未知哪种启动子将最适于在人t细胞中表达。为了比较启动子,用从各种启动子表达绿色荧光蛋白(gfp)的慢病毒载体转导肾细胞系和树突细胞系。发现人ef1α启动子在树突细胞系bdcm中是最强启动子(图1)。该启动子在肾细胞系293t中也非常强。使用biogps.org上的数据组分析各种启动子的细胞特异性表达(图2)。从该数据组中,ef1α启动子在t细胞中的表达与在bdca+细胞中的表达相似。事实上,先前使用人ef1α启动子在活化的人t细胞中表达car。budde等,plos one 8(12):e82742(2013)。
63.基于不同细胞类型中表达的差异,哪种启动子可成功地用于在人t细胞中表达car不是显而易见的。评价人泛素mhc i,mhc ii,和β2微球蛋白(β2m)启动子在人t细胞中表达car的适用性。意外的是,发现所有这些启动子在人t细胞中均发挥作用(图3a
‑
c)。
64.为了发现生成car t
‑
细胞的最佳条件,测试了几种可能影响t细胞转导和car表达的参数(例如,moi、孵育时间、启动子、t细胞活化和纯化策略)。
65.在减去背景(未转导的(utd)对照中car+细胞的%)之后,在24小时(moi=10和30的ubc载体)、96小时(moi=3的β2m载体以及moi=3和10的ubc载体)和第8天(moi=30的β2m载体和每个测试的moi的ubc载体)检测car+细胞。在96小时和第8天,发现与未转导的淋巴细胞相比高百分比(约30%)的car
‑
cd19阳性细胞。在第7天,当分析活cd3+群时,发现与未转导的淋巴细胞相比高百分比(约70%)的car
‑
cd19阳性细胞。
66.发现在几个供体中car
‑
cd19的强表达(高至cd3阳性淋巴细胞的70%)。甚至在moi=3下发现car表达;然而,在较晚时间点(在该实验中第7天)实现了高百分比的car
‑
阳性细胞。有趣的是,供体之间的car表达是可变的。对于car
‑
cd19,表达限于cd3阳性群,因为cd3阴性细胞无法表达car
‑
cd19。
67.hook
‑
链霉亲和素序列被克隆到以与vsv
‑
g融合的形式编码密码子优化的vsg
‑
g蛋白的假性载体中。通过与编码gfp的慢病毒共转染和hook
‑
链霉亲和素编码载体的剂量递增来评价该载体。未见由于er中vsv蛋白质的停留而导致的效价降低(图4)。免疫荧光研究显示在er中没有vsv蛋白质的停留。这与单独vsv蛋白质所显示的结果相反。nature methods 9(5):493
‑
500(2102)。因此,不清楚hook
‑
链霉亲和素系统是否可调控car表达。
68.评价胞内结构域之间各种位置上用链霉亲和素结合蛋白(sbp)在t细胞的表面上表达car
‑
cd19或car
‑
cd19以检查sbp序列的存在是否修饰car表达。具有各种位置sbp的car
‑
cd19+被克隆到β2m启动子控制下的慢病毒载体中。
69.用慢病毒载体转导人t细胞并且评价细胞的表面上的cd19的表达。car
‑
cd19
‑
sbp
在t细胞的表面上表达,与car
‑
cd19相比,mfi轻微降低(图5)。
70.在hook
‑
链霉亲和素存在下,评价具有胞内结构域间不同位置的sbp的car
‑
cd19慢病毒载体。用表达car
‑
cd19
‑
sbp的慢病毒载体和表达hook
‑
链霉亲和素的慢病毒载体共转导人t
‑
细胞并且在添加生物素之前或之后评价转导的细胞的百分比和mfi。发现hook
‑
链霉亲和素和car
‑
cd19
‑
sbp可在人t
‑
细胞中共表达。hook
‑
链霉亲和素的存在可保留t
‑
细胞内质网中的car
‑
cd19
‑
sbp并且在培养基中添加生物素诱导car
‑
cd19释放及其在细胞表面上的表达(图6)。
71.然后构建各种慢病毒载体含有hook和在各种位置含有sbp的car(图11)。构建体含有与hook链霉亲和素连接的β2m启动子,之后是与具有3个不同位置的sbp的car
‑
cd19连接的ires。
72.用慢病毒载体转导来自各种供体的人t
‑
细胞并且在生物素存在和缺失下在细胞表面上评价car
‑
cd19
‑
sbp的表达。在人t
‑
细胞中表达hook
‑
ires
‑
car
‑
cd19
‑
sbp。hook
‑
链霉亲和素的表达可保留t
‑
细胞的内质网中的car
‑
cd19
‑
sbp,无论其位置。在培养基中添加生物素诱导car
‑
cd19的释放和其在细胞表面上的表达,与其位置无关(图7)。
73.本发明包括编码car的慢病毒载体,及其在宿主,尤其是人中诱导免疫应答的用途。
74.在公开和描述本发明的蛋白质、组合物、方法和其他实施方式之前,应理解本文的术语只是用于描述具体实施方式的目的而并非旨在进行限制。必须指出,除非上下文另有明确说明,否则在本说明书和所附权利要求书中使用的单数形式的“一个”“、一种”和“该”包括复数的指代对象。
75.本文所用的术语“包含”与“包括”或“含有”同义,为封闭式或开放式表述,并不排除其它未列举的要素或方法步骤。
76.本文中氨基酸的全名可与各自的标准三字母和单字母缩写互换使用。为避免疑虑,那些是:丙氨酸(ala,a),精氨酸(arg,r),天冬酰胺(asn,n),天冬氨酸(asp,d),半胱氨酸(cys,c),谷氨酸(glu,e),谷氨酰胺(gln,q),甘氨酸(gly,g),组氨酸(his,h),异亮氨酸(ile,i),亮氨酸(leu,l),赖氨酸(lys,k),甲硫氨酸(met,m),苯丙氨酸(phe,f),脯氨酸(pro,p),丝氨酸(ser,s),苏氨酸(thr,t),色氨酸(trp,w),酪氨酸(tyr,y),缬氨酸(val,v)。
77.本文所用术语“体外”指在人工环境中(例如在试管或反应容器中、在细胞培养物中、在培养皿中等),而非在生物体(例如动物、植物或微生物)内发生的事件。
78.本文所用术语“体内”指在生物体(例如动物、植物或微生物)内发生的事件。
79.本文所有术语“分离的”指:(1)已与其最初形成时(天然或实验环境中)相关的至少一些组分分离的物质或实体,和(2)通过人工生成、制备和/或制造的物质或实体。分离的物质和/或实体可与至少约10%、约20%、约30%、约40%、约50%、约60%、约70%、约80%、约90%或更多的其最初相关的其他组分分离。在一些实施方式中,分离的实际是约80%、约85%、约90%、约91%、约92%、约93%、约94%、约95%、约96%、约97%、约98%、约99%,或超过约99%纯。如本文中所用,如果某物质基本不含其他组分,则认为其是“纯”的。
80.本发明的“分离的”产物,包括分离的核酸、蛋白质、多肽和抗体不是天然产物(即,“非天然产生”)。相反,本发明的“分离的”核酸、蛋白质、多肽和抗体是“人工”产物。本发明
的“分离的”产物可与天然产物“显著不同”或“明显不同”。作为非限制性示例,分离的核酸可经纯化、重组、合成、标记、和/或附连至固体基材。这类核酸可与天然产生的核酸显著不同或明显不同。作为其他非限制性示例,本发明的“分离的”蛋白质、多肽和抗体可经纯化、重组、合成、标记、和/或附连至固体基材。这类蛋白质、多肽和抗体可与天然产生的蛋白质、多肽和抗体显著不同或明显不同。
81.本文所用术语“肽”指短的多肽,例如其通常含有少于约50个氨基酸且更通常含有少于约30个氨基酸。本文中的该术语包括模拟结构和生物学功能的类似物和模拟物。
82.术语“多肽”包括天然产生和非天然产生的蛋白质、其片段、突变体、衍生物和类似物。多肽可以是单聚体或多具体。此外,多肽可包含多个不同结构域,其中各结构域具有一种或多种不同活性。为避免疑问,“多肽”可以是超过两个氨基酸的任何长度。
83.术语“分离的蛋白质”或“分离的多肽”指特定的蛋白质或多肽,从本义或派生含义上说,其(1)与天然状态下伴随其的天然相关组分不相关,(2)以天然中未发现的纯物质形式存在,其中纯物质可以针对其他细胞材料的存在来判定(例如不含来自相同物种的其他蛋白质),(3)由来自其他物种的细胞表达,或(4)不存在于天然中(例如,其是天然中发现的多肽的片段或者其包含天然中未发现的氨基酸类似物或衍生物或者不同于标准肽键的连接)。因此,化学合成或者在与其天然来源细胞不同的细胞系统中合成的多肽是与其天然相关组分“分离的”。使用本领域已知的蛋白质纯化技术,还可通过分离使多肽或蛋白质基本不含天然相关的组分。如本文所定义,“分离的”无需将所述蛋白质、多肽、肽或寡肽从其合成所在的细胞中物理移出。
84.可纯化蛋白质或多肽。优选地,纯化的蛋白质或多肽的纯度超过50%、75%、85%、90%、95%、97%、98%、或99%。在本发明的内容中,纯度超过50%(等)的纯化的蛋白质表示纯化的蛋白质样品含有少于50%(等)的其他蛋白质。例如,如果包含蛋白质的样品含有少于1%的污染宿主细胞蛋白质,则其纯度可以是99%。
85.本文所用术语“多肽片段”指具有缺失的多肽,例如与全长多肽(如天然产生的蛋白质)相比缺失氨基末端和/或羧基末端。在一个实施方式中,该多肽片段是连续序列,其中片段的氨基酸序列与天然产生的序列中相应位置相同。片段长度通常是至少5、6、7、8、9或10个氨基酸,或至少12、14、16或18个氨基酸,或至少20个氨基酸,或至少25、30、35、40或45个氨基酸,或至少50或60个氨基酸,或至少70个氨基酸,或至少100个氨基酸。
86.术语“融合蛋白”指包含与异源氨基酸序列偶联的多肽或片段的多肽。融合蛋白是有用的,因为可对其进行构建以使其含有两个或更多个所需功能元件,所述功能元件可来自两种或更多种不同蛋白质。融合蛋白包含来自感兴趣多肽的至少10个连续氨基酸,或至少20或30个氨基酸,或至少40、50或60个氨基酸,或至少75、100或125个氨基酸。融合蛋白中包含的异源多肽长度通常是至少6个氨基酸,或至少8个氨基酸,或至少15、20或25个氨基酸。特别有用的是包含较大多肽的融合体,如igg fc区,甚至整个蛋白质,如含有绿色荧光蛋白(“gfp”)生色团的蛋白质。通过构建与编码不同蛋白质或肽的核酸序列框内融合的编码多肽或其片段的核酸序列并随后表达该融合蛋白,可重组生成融合蛋白。或者,可通过将多肽或其片段与另一蛋白质交联来化学生成融合蛋白。
87.本文所用的“重组”可指生物分子,例如,基因或蛋白质,或指代生物体。术语“重组”可用于克隆的dna分离物、化学合成的多核苷酸、或由异源系统生物合成的多核苷酸、以
及由这类核酸编码的蛋白质或多肽和/或rna。“重组”核酸是与自然中其不连接的核苷酸或多核苷酸连接的核酸。“重组”蛋白质或多肽可以是(1)与自然中其不连接的氨基酸或多肽连接的蛋白质或多肽;和/或(2)通过重组核酸的转录和/或翻译产生的蛋白质或多肽。因此,由微生物合成的蛋白质是重组的,例如,如果其从由细胞中存在的重组核酸合成的mrna合成。“重组”细胞是包含“重组”生物分子的细胞。例如,包含“重组”核酸的t细胞是“重组”细胞。
88.术语“多核苷酸”、“核酸分子”、“核酸”、或“核酸序列”是指长度为至少10个碱基的核苷酸的聚合形式。该术语包括dna分子(例如,edna或基因组或合成dna)和rna分子(例如,mrna或合成rna),以及含有非天然核苷酸类似物、非天然核苷间键或两者的dna或rna的类似物。核酸可以是任何拓扑构型。例如,核酸可以是单链、双链、三链、四聚体、部分双链、分支、发夹形、环形或锁式构型。该核酸(也称为多核苷酸)可包括rna、edna、基因组dna、以及上述的合成形式和混合聚合物的正义链和反义链。它们可经化学或生物修饰或者可含有非天然或衍生的核苷酸碱基,如本领域技术人员将易于想到的那样。这类修饰包括,例如,标记物,甲基化,用类似物取代一个或多个天然产生的核苷酸,核苷酸间修饰如不带电连接(例如,膦酸甲酯、磷酸三酯、氨基磷酸酯、氨基甲酸酯等),带电连接(例如,硫代磷酸酯、二硫代磷酸酯等),侧接部分(例如,多肽),嵌入剂(例如,吖啶、补骨脂素等),螯合剂,烷化剂,和修饰的连接(例如,α异头核酸等)。还包括模拟多核苷酸通过氢键和其他化学相互作用结合至指定序列的合成分子。这类分子是本领域已知的并且包括,例如,其中肽连接取代分子主链中磷酸酯连接的那些。其他修饰可包括,例如,类似物,其中核糖环含有桥连部分或其他结构,如在“锁”核酸中发现的修饰。
[0089]“合成”rna、dna或混合聚合是在细胞外产生的,例如,化学合成的。
[0090]
本文所用术语“核酸片段”是指与全长参考核苷酸序列相比具有缺失,例如,5
’‑
末端或3
’‑
末端缺失的核酸序列。在一个实施方式中,该核酸片段是连续序列,其中片段的核苷酸序列与天然产生的序列中相应位置相同。在一些实施方式中,片段长度为至少10、15、20、或25个核苷酸,或至少20、30、40、50、60、70、80、90、100、110、120、130、140、或150个核苷酸。在一些实施方式中和,核酸序列片段是开放阅读框序列片段。在一些实施方式中,该片段编码由开放阅读框核苷酸序列编码的蛋白质的多肽片段(如本文定义)。
[0091]
可纯化核酸。优选地,纯化的核酸的纯度超过50%、75%、85%、90%、95%、97%、98%、或99%。在本发明的范围内,纯度至少是50%的纯化的核酸表示纯化的核酸含有少于50%的其他核酸。例如,如果质粒样品含有少于1%的污染细菌dna,则其纯度可以是至少99%。
[0092]
如本文所用,生物体的基因组中的内源性核酸序列(或该序列的编码蛋白质产物)在本文中被认为是“重组的”,如果将异源序列与内源性核酸序列相邻设置。在本文中,异源序列是天然不与内源性核酸序列相邻的序列,无论异源序列是自身内源性(来自同一宿主细胞或其后代)或外源性(来自不同宿主细胞或其后代)。例如,宿主细胞的基因组中基因的天然启动子可被启动子序列取代(例如,通过同源重组),使得该基因具有改变的表达模式。该基因现在将变得“重组”,因为其与天然与其侧接的序列中的至少一些分开。
[0093]
如果核酸含有基因组中相应核酸不天然产生的任何修饰,则该核酸也被认为是“重组”的。例如,如果内源性编码序列(例如,通过人介入)含有人工导入的插入、缺失或点
突变,则该内源性编码序列被认为“重组”的。“重组核酸”也包括在异源位点整合到宿主细胞染色体的核酸和以原生质体存在的核酸构建体。
[0094]
本文所用术语参考核酸序列的“简并变体”包括可按照标准遗传代码被翻译以提供与从参考核酸序列翻译的相同的氨基酸序列的核酸序列。使用术语“简并寡核苷酸”或“简并引物”来表示能够与靶核酸序列杂交的寡核苷酸,其不必在序列上相同但是其在一个或多个特定区段内互相同源。
[0095]
用于指代核酸序列时,术语“序列相同性百分数”或“相同性”指以最大对应性进行比对时两条序列中相同的残基。序列相同性比较的长度可超过一段至少约9个核苷酸,通常至少约20个核苷酸,共通常至少约24个核苷酸,一般至少约28个核苷酸,共一般至少约32,并且甚至更一般至少约36个或更多个核苷酸。存在多种本领域已知的不同算法,其可用于测量核苷酸序列相同性。例如,fasta、gap或bestfit来比较多核苷酸序列,它们是威斯康星州麦迪逊的遗传计算机组(gcg)威斯康星包版本10.0中的程序。fasta可提供查询序列与检索序列之间最佳重叠区域的比对和序列相同性百分比。pearson,methods enzymol.183:63
‑
98(1990)。例如,可使用gcg 6.1版(通过引用纳入本文)提供的默认参数的fasta(字长为6和用于计分矩阵的nopam系数)或使用默认参数的gap来确定核酸序列之间的序列相同性百分比。或者,可使用计算机程序,blast(altschul等,j.mol.biol.215:403
‑
410(1990);gish和states,nature genet.3:266
‑
272(1993);madden等,meth.enzymol.266:131
‑
141(1996);altschul等,nucleic acids res.25:3389
‑
3402(1997);zhang和madden,genome res.7:649
‑
656(1997)),特别是blastp或tblastn(altschul等,nucleic acids res.25:3389
‑
3402(1997))比较序列。
[0096]
本文所用“表达控制序列”是指影响它们可操作连接的编码序列的表达的多核苷酸序列。表达控制序列是控制核酸序列的转录、转录后事件和翻译的序列。表达控制序列包括合适的转录起始、终止、启动子和增强子序列;有效的rna加工信号例如剪接和聚腺苷酸化信号;稳定细胞质mrna的序列;提高翻译效率的序列(例如,核糖体结合位点);增强蛋白质稳定性的序列;以及在需要时增强蛋白质分泌的序列。这类控制序列的性质根据宿主生物素而不同;在原核生物中,这类控制序列一般包括启动子、核糖体结合位点、和转录终止序列。术语“控制序列”旨在最低限度包括其存在对表达必要的所有组分,且还可包括其存在是有益的其他组分,例如前导序列和融合伙伴序列。
[0097]
本文所用的“可操作连接的”或“可操作地连接的”表达控制序列是指其中表达控制序列与感兴趣基因连续以控制感兴趣基因,以及表达控制序列以反式或在一定距离处发挥作用以控制感兴趣基因的连接。
[0098]
如本文所用,“载体”旨在指代能够运输所连接的另一核酸的核酸分子。一种类型的载体是“质粒”,其一般是指环形双链dna环,其中可连接其他dna区段,但也包括线性双链分子,如从通过聚合酶链反应(pcr)扩增或用限制性酶处理环形质粒所得的那些。其他载体包括粘粒、细菌人工染色体(bac)和酵母人工染色体(yac)。载体的另一类型是病毒载体,其中可将额外dna区段连接进入病毒基因组(在下文中详述)。某些载体能够在其转导的宿主细胞中自主复制(例如,具有在宿主细胞中发挥功能的复制起点的载体)。可在导入宿主细胞后将其他载体整合到宿主细胞的基因组中,从而与宿主基因组一起复制。此外,某些载体能够引导与其操作性连接的基因的表达。这类载体在本文中称为“重组表达载体”(或简称
“
表达载体”)。整合粘粒载体pyub412是“载体”的一个示例。
[0099]
本文所用术语“重组宿主细胞”(或简称“重组细胞”或“宿主细胞”)旨在指代其中已导入重组核酸例如重组载体的细胞。在一些情况中,用说明细胞类型的名称取代词“细胞”。例如,“重组微生物”是重组宿主细胞,其是微生物宿主细胞。应理解,所述术语不仅指具体对象细胞,还指所述细胞的后代。由于在连续传代过程中可能因突变或环境影响而发生某些改变,这类后代事实上可能不与亲代细胞相同,但仍落入本文所用术语“重组宿主细胞”、“重组细胞”和“宿主细胞”的范围内。重组宿主细胞可以是在培养中生长的分离的细胞或细胞系或者可以是位于活组织或生物体中的细胞。
[0100]
本文所用术语“哺乳动物”是指哺乳动物纲的任何成员,包括胎盘哺乳动物和有袋类哺乳动物。因此,“哺乳动物”包括人、灵长类、牲畜和实验室哺乳动物。示例性的哺乳动物包括啮齿类、小鼠、大鼠、兔、狗、猫、绵羊、马、山羊、美洲驼、牛、灵长类、猪、和任何其他哺乳动物。在一些实施方式中,哺乳动物是转基因哺乳动物、遗传工程改造的哺乳动物和克隆的哺乳动物中的至少一种。
[0101]
嵌合抗原受体
[0102]
本发明包括嵌合抗原受体(car),其包含胞外抗原结合结构域(结合结构域)、铰链和跨膜结构域(跨膜结构域)、hook
‑
结合结构域、和胞内信号转导结构域(活化结构域)。car可含有这些各结构域中的1个、2个、3个或更多个。
[0103]
本发明单独包括本文应用的特定多肽及其片段的全部可能组合。
[0104]
本发明包括包含hook
‑
结合结构域的car。“hook
‑
结合结构域”是可逆地直接或间接结合至细胞内的“hook”蛋白的结构域,其结合防止car在合适条件下离开内质网(er)或高尔基体。
[0105]
在一个实施方式中,hook
‑
结合结构域包括链霉亲和素
‑
结合肽(sbp),其可结合至含有核链霉亲和素的hook蛋白。生物素通过与sbp竞争使含有钩
‑
结合结构域的car从hook上释放。然后car可移向细胞膜。
[0106]
优选地,可采用称为rush(使用选择性hook的停留)系统的系统,boncompain等,nat.methods 9:493
‑
498,2012,其通过引用纳入本文。
[0107]
优选地,hook
‑
结合结构域包括以下氨基酸序列:
[0108]
mdekttgwrgghvveglageleqlrarlehhpqgqrep(seq id no:1)或者由以下核酸序列编码:
[0109][0110]
可以相同功效使用在其n
‑
末端和c
‑
末端处缺失的较短sbp片段。参见barrette
‑
ng,i.h.,s.c.wu,w.m.tjia,s.l.wong和k.k.ng.2013,sbp
‑
标签
‑
链霉亲和素复合物的结构显示在分开的亚基上的新螺旋折叠结合袋(the structure of the sbp
‑
tag
‑
streptavidin complex reveals a novel helical scaffold bridging binding pockets on separate subunits),acta crystallographica.d部分,biological crystallography 69:879
‑
887。
[0111]
hook
‑
结合结构域的一个实施方式示于wo2010/142785,其通过引用纳入本文。fkbpf
‑
k506结合结构域12(fkbpi2)可与用作hook的fkbp
‑
雷帕霉素相关蛋白(frap)联用。在该实施方式中,相互作用仅在雷帕霉素或其类似物存在下发生,其能够介导fkbp12和frap之间的相互作用,并且具体可选自fki012、fk
‑
csa和雷帕霉素。
[0112]
在一个实施方式中,hook
‑
结合结构域位于car的胞质内区域中。在其他实施方式中,hook
‑
结合结构域位于其他位置,即,完全在不同胞质内元件之间(例如,跨膜区域和第一共刺激元件)或不同同刺激元件之间的连接。
[0113]
本发明包括含有结合结构域的car,该结合结构域包含特异性结合至人多肽的抗体。术语“抗体”包括多克隆抗体、单克隆抗体、其片段如f(ab
′
)2和fab片段、单链可变片段(scfv)、单结构域抗体片段(vhh或纳米抗体,优选骆驼科)、以及二价和三价抗体片段(双抗体和三抗体)。
[0114]
优选地,抗体是单链fv抗体或纳米抗体。
[0115]
在一个实施方式中,抗体是单特异性的。在一个实施方式中,抗体对于2、3、或4种多肽是多特异性的。优选地,抗体是双特异性的。
[0116]
抗体可以是合成、单克隆或多克隆的并且可由本领域熟知的技术制备。这类抗体通过抗体的抗原结合位点特异性结合至人蛋白质(与非特异性结合相反)。人蛋白质、多肽片段和肽在产生与之有免疫反应性的抗体中可用作免疫原。人蛋白质、多肽和肽含有抗原决定簇或表位,其引发抗体形成。
[0117]
这些抗原决定簇或表位可以是线性或构象的(不连续)。线性表位包含多肽的氨基酸的单区段,而构象或不连续表位包含来自多肽的不同区域的氨基酸区段,其在蛋白质折叠之后密切相邻(c.a.janeway,jr.和p.travers,immuno biology 3:9(加兰出版公司(garland publishing inc.),第二版,1996))。由于折叠的蛋白质具有复杂表面,可及的表位数量是非常多的;然而,由于蛋白质构象和空间位阻,实际结合至表位的抗体数量少于可及表位的数量(c.a.janeway,jr.和p.travers,immuno biology 2:14(加兰出版公司,第二版,1996))。可通过任意本领域已知的方法来鉴定表位。
[0118]
因此,本发明的一个方面涉及人蛋白质的抗原性表位。这类表位可用于产生抗体,尤其是单克隆抗体,如下文详述。
[0119]
如果抗体以大于或等于约107m
‑1的ka结合人蛋白质或多肽,则它们被定义为特异性结合。可易于使用常规技术,例如,scatchard等,ann.n.y.acad.sci.,51:660(1949)所述的那些确定结合伴侣或抗体的亲和性。
[0120]
使用本领域熟知的方法可易于从多种来源生成多克隆抗体,例如,马、牛、山羊、绵羊、狗、鸡、兔、小鼠、或大鼠。一般而言,一般通过胃肠外注射向宿主动物给予适当偶联的纯化的人蛋白质或多肽。可通过使用佐剂来增强免疫原性,例如,完全或不完全的弗氏佐剂。在加强免疫之后,小血清样品经收集并测试针对人蛋白质或多肽的反应性。可用于这种确定的各种试验的示例包括《抗体:实验室手册》(antibodies:a laboratory manual),harlow和lane编写,冷泉港实验室出版社,1988中所述的那些;以及过程,如逆流免疫电泳(ciep)、放射免疫试验、放射
‑
免疫沉淀、酶联免疫吸附试验(elisa)、点印迹试验、和夹心试验。参见美国专利号4,376,110和4,486,530。
[0121]
使用熟知的方法能够容易地制备单克隆抗体。参见,例如,美国专利号re 32,011、
4,902,614、4,543,439、和4,411,993中所述的过程;《单克隆抗体,杂交瘤:生物学分析的新领域》(monoclonal antibodies,hybridomas:a new dimension in biological analyses),普莱努出版公司,kennett、mckeam和bechtol编写,1980。
[0122]
例如,宿主动物,如小鼠,可腹膜内注射至少一次,并且约3周间隔注射至少两次分离并纯化的人蛋白质或偶联的人多肽,例如,包含或由上述特定氨基酸组成的肽。然后通过常规点印迹技术或抗体捕获(abc)来测试小鼠血清以确定哪种动物最佳融合。大约2
‑
3周后,向小鼠给予人蛋白质或多肽的静脉内加强。之后,处死小鼠,并且脾细胞按照成熟方案与市售骨髓瘤细胞如ag8.653(atcc)融合。简言之,骨髓瘤细胞在培养基中洗涤数次并以约3个脾细胞:1个骨髓瘤细胞的比例融合至小鼠脾细胞。融合剂可以是本领域使用的任意合适试剂,例如,聚乙二醇(peg)。融合物接种到含有培养基的平板上以允许融合的细胞的选择性生长。然后可使融合的细胞生长约8天。来自所得杂交瘤的上清经收集并加入用山羊抗小鼠ig首次包被的平板。洗涤后,将标记物,如带标记的人蛋白质或多肽,加入各孔并孵育。可随后检测阳性孔。阳性克隆在大培养物中生长并且随后在蛋白a柱(法玛西亚公司)上纯化上清。
[0123]
可使用其他技术来产生本发明的单克隆抗体,如alting
‑
mees等,
″
单克隆抗体表达文库:杂交瘤的快速替代物(monoclonal antibody expression libraries:a rapid alternative to hybridomas)
″
,strategies in molecular biology 3:1
‑
9(1990)所述的那些,其通过引用纳入本文。类似地,可使用重组dna技术来构建结合伴侣以纳入编码特定结合抗体的基因可变区。这种技术描述于larrick等,biotechnology,7:394(1989)。
[0124]
本发明也包括可通过常规技术产生的这类抗体的结合片段。这类片段的示例包括但不限于fab和f(ab
′
)2片段。也提供由遗传工程改造技术产生的抗体片段和衍生物。
[0125]
本发明的单克隆抗体包括嵌合抗体,例如,鼠单克隆抗体的人源化形式。这类人源化抗体可通过已知技术制备,并且在抗体给予人时提供降低的免疫原性的优势。在一个实施方式中,人源化的单克隆抗体包含鼠抗体的可变区(或其抗原结合位点)和衍生自人抗体的恒定区。或者,人源化的抗体片段可包含鼠单克隆抗体的抗原结合位点和衍生自人抗体的可变区片段(缺少抗原结合位点)。用于产生嵌合并进一步工程改造的单克隆抗体的过程包括riechmann等(nature 332:323,1988),liu等(pnas 84:3439,1987),larrick等(bio/technology 7:934,1989),以及winter和harris(tips 14:139,1993年5月)中所述的那些。通过转基因生成抗体的过程可发现于gb 2,272,440、美国专利号5,569,825和5,545,806。
[0126]
可使用通过遗传工程改造方法产生的抗体,如嵌合和人源化单克隆抗体,同时包含人和非人部分,其可使用标准重组dna技术制备。可通过使用本领域已知的标准dna技术的遗传工程改造来产生这类嵌合和人源化单克隆抗体,例如,使用robinson等,国际公开号wo 87/02671;akira等,欧洲专利申请0184187;taniguchi,m.,欧洲专利申请0171496;morrison等,欧洲专利申请0173494;neuberger等,pct国际公开号wo 86/01533;cabilly等,美国专利号4,816,567;cabilly等,欧洲专利申请0125023;better等,science 240:1041 1043,1988;liu等,pnas 84:34393443,1987;liu等,j.immunol.139:35213526,1987;sun等,pnas 84:214 218,1987;nishimura等,canc.res.47:9991005,1987;wood等,nature 314:446 449,1985;和shaw等,j.natl.cancer inst.80:1553 1559,1988);morrison,s.l.,science 229:1202 1207,1985;oi等,biotechniques 4:214,1986;winter,美国专利
号5,225,539;jones等,nature 321:552 525,1986;verhoeyan等,science 239:1534,1988;和beidler等,j.immunol.141:4053 4060,1988中所述的方法。
[0127]
可由展示包装体群(优选衍生自丝状噬菌体)表达免疫球蛋白文库,以形成抗体展示文库。特别适用于生成抗体展示文库的方法和试剂的示例可见于,例如,ladner等,美国专利号5,223,409;kang等,pct公开号wo 92/18619;dower等,pct公开号wo 91/17271;winter等,pct公开号wo 92/20791;markland等,pct公开号wo 92/15679;breitling等,pct公开号wo 93/01288;mccafferty等,pct公开号wo 92/01047;garrard等,pct公开号wo 92/09690;ladner等,pct公开号wo 90/02809;fuchs等,(1991)bio/technology 9:1370 1372;hay等,(1992)hum antibod hybridomas 3:81 85;huse等,(1989)science 246:1275 1281;griffths等,(1993)同上;hawkins等,(1992)j mol biol 226:889 896;clackson等,(1991)nature 352:624 628;gram等,(1992)pnas 89:3576 3580;garrad等,(1991)bio/technology 9:1373 1377;hoogenboom等,(1991)nuc acid res 19:41334137;和barbas等,(1991)pnas 88:7978 7982。一旦在展示包装物(例如,丝状噬菌体)表面上展示,分泌抗体文库以鉴定并分离表达结合人蛋白质或多肽的抗体的包装物。在优选的实施方式中,文库的初步筛选包括用固定的人蛋白质或多肽淘选,并且选择表达展示结合固定的人蛋白质或多肽的展示包装物。
[0128]
与合成和半合成抗体联用时,这类术语往往涵盖但不限于抗体片段、同种型交换的抗体、人源化抗体(例如,小鼠
‑
人,人
‑
小鼠)、杂交体、具有多种特异性的抗体、和全合成抗体
‑
样分子。
[0129]
本发明包括包含cd3
‑
ζ或fc受体γ氨基酸序列的活化结构域的car。sadelain等,cancer discov.2013年4月;3(4):388
‑
398,其通过引用纳入本文。本发明还包括car,其包括包含cd3
‑
ζ链和共刺激受体如cd28、4
‑
1bb(cd137)、dap10、ox40(cd134)、icos、cd27、或cd40l的胞质结构域的活化结构域的car。同上。
[0130]
优选地,car包含以下具有t
‑
细胞活化活性的氨基酸序列中至少一种的至少50、60、70、80、90、100、110、120、150、或200个氨基酸的片段。
[0131]
cd3
‑
ζ:
[0132][0133]
cd28:
[0134][0135]4‑
1bb(cd137):
[0136][0136][0137]
dap10:
[0138][0139]
ox40(cd134):
[0140][0141]
icos:
[0142]
[0143]
cd27:
[0144][0145]
cd40l(cd154):
[0146][0147]
在各种实施方式中,car包含至少20、30、40、50、60、70、80、90、100、110、120、150、或200个氨基酸的片段,其与seq id no:3、seq id no:4、seq id no:5、seq id no:6、seq id no:7、seq id no:8、seq id no:33、或seq id no:34的氨基酸序列具有至少90%、优选超过95%、更优选超过99%的相同性。
[0148]
在各种实施方式中,car的活化结构域包含至少20、30、40、50、60、70、80、90、100、110、120、150、或200个氨基酸的1、2、3、或4个片段,其与seq id no:3、seq id no:4、seq id no:5、seq id no:6、seq id no:7、seq id no:8、seq id no:33、或seq id no:34的氨基酸序列具有至少90%、优选超过95%、更优选超过99%的相同性。
[0149]
本发明包括car,其包含跨膜(tm)结构域,优选至少20、30、40、50、60、70、80、90、100、110、120、150、或200个氨基酸的片段,最优选cd28、cd3z、cd8、cd4、fcrγ、tm区域中至少一个的片段。
[0150]
car可经纯化。优选地,纯化的car的纯度超过50%、75%、85%、90%、95%、97%、98%、或99%。在本发明的内容中,纯度超过50%(等)的纯化的car表示纯化的car样品含有少于50%(等)的其他蛋白质。例如,如果从宿主细胞纯化的重组car的样品含有少于1%的污染宿主细胞蛋白质,则其纯度可以是99%。
[0151]
在一个优选的实施方式中,car编码seq id no:46、seq id no:48、或seq id no:50中任一个的氨基酸序列。
[0152]
特别优选的car包括编码由图11中所示的car的任意组分的那些,包括信号序列、cd19 scfv、cd8铰链、跨膜结构域、4
‑
1bb、和/或cd3z序列。其他优选的car包括编码seq id no:44
‑
50的car的任意组分的那些。特别优选的car具有由另一个结合区域替代的cd19 scfv结构域。
[0153]
核酸
[0154]
本发明包括编码car的核酸。核酸可以是单链或双链的。核酸可以是rna或dna分子。优选的核酸编码本文详述的seq id no中至少一个的氨基酸序列。本发明包括插入载体的本发明的分离的核酸。
[0155]
在一个实施方式中,核酸序列包括seq id no:2的核酸序列。在一个实施方式中,核酸序列包括以下核酸序列中的一个或多个:
[0156]
hook:(核+ha标签+其他结构)
[0157][0157]
[0158]
核心链霉亲和素:
[0159][0160]
ha标签:tatccgtacgacgtaccagactacgca(seq id no:37)。
[0161]
优选的核酸是至少50、60、70、80、90、100、110、120、150、200、300、400、500、或600个核苷酸。
[0162]
可纯化核酸。优选地,纯化的核酸的纯度超过50%、75%、85%、90%、95%、97%、98%、或99%。在本发明的范围内,纯度超过50%的纯化的核酸表示纯化的核酸含有少于50%的其他核酸。例如,如果从宿主细菌纯化的质粒样品含有少于1%的污染细菌dna,则其纯度可以是至少99%。
[0163]
特别优选的核酸包括以下:
[0164]
car_cd19第二代:1518bp
[0165]
[0166]
car_cd19第三代:1641bp
[0167]
[0168]
特别优选的核酸包括包含或编码由图9中所示的car的任意组分的那些,包括信号序列、cd19 scfv、cd8铰链、跨膜结构域、4
‑
1bb、和/或cd3z序列。其他优选的核酸包括包含或编码seq id no:45
‑
50的car的任意组分的那些。
[0169]
在一些实施方式中,核酸包含可操作地连接至启动子,优选ubc或β2m的hook,和包含可操作地连接至ires的hook
‑
结合蛋白的car。在优选的实施方式中,hook是链霉亲和素蛋白,优选核心链霉亲和素,并且hook
‑
结合蛋白是链霉亲和素
‑
结合蛋白。
[0170]
载体
[0171]
本发明包括编码car和hook
‑
结合结构域的载体。优选的载体包括核酸序列或编码本文详述的seq id no中至少一个的氨基酸序列。
[0172]
载体还可编码“hook”。“hook”是防止含hook
‑
结合结构域的car通过直接或间接可逆结合car内的hook
‑
结合结构域而离开内质网(er)或高尔基体。
[0173]
停留可发生在er的腔中或在其胞质面处,这取决于蛋白质的拓扑异构学和相互作用结构域触发的取向。boncompain等,《新编细胞生物学方案》(current protocols in cell biology)15.19.1
‑
15.19.16,2012年12月,其通过引用纳入本文。
[0174]
在一些实施方式中,er的hook包含基质相互作用分子1(stim1
‑
nn;i型蛋白质)的突变体,其位于er内但可能不结合微管,具有n
‑
末端精氨酸基基序的主要阻止相容性复合物(ii;ii型蛋白质)的人非变异链的同种型;或c
‑
末端er滞留信号(lys
‑
asp
‑
glu
‑
leu;kdel)。boncompain等,nat.methods 9:493
‑
498,2012,其通过引用纳入本文。hook可与核心链霉亲和素在其腔或胞质结构域中融合,取决于hook
‑
结合蛋白质,同上,图1。
[0175]
在替代性实施方式中,针对高尔基体的hook可以是golgin
‑
84,其被用作胞质高尔基体hook。同上。
[0176]
优选地,该hook包含链霉亲和素蛋白质序列,最优选核心链霉亲和素。美国专利号5,672,691,其通过引用纳入本文。
[0177]
优选地,该hook包含以下链霉亲和素蛋白质序列之一:
[0178][0178]
或
[0179][0179]
或
[0180][0180][0181]
优选地,由以下核苷酸序列编码hook:
[0182][0182][0183]
在一些实施方式中,seq id no:31或seq id no:32的氨基酸49处的甘氨酸被大体积残基(例如,苏氨酸)取代以降低生物素结合亲和性但不影响sbp结合亲和性。wu等,plos one 8(7):e69530(2013),其通过引用纳入本文。也可导入另一个突变来使sbp结合以进一步有利于生物素(突变s27a)。
[0184]
在一些实施方式中,载体包含可操作地连接至启动子,优选ubc或β2m的hook,和包含可操作地连接至ires的hook
‑
结合蛋白的car。
[0185]
优选的ires核苷酸序列是:
[0186][0187]
在优选的实施方式中,hook是链霉亲和素蛋白,优选核心链霉亲和素,并且hook
‑
结合蛋白是链霉亲和素
‑
结合蛋白。优选的载体是包含以下核苷酸或氨基酸序列中任一个的慢病毒载体:
[0188]
car
‑
cd19第二代
‑
sbp1(nt):
[0189]
[0190]
[0191]
car
‑
cd19第二代
‑
sbp1(aa):
[0192][0193]
car
‑
cd19第二代
‑
sbp2(nt):
[0194]
[0195]
car
‑
cd19第二代
‑
sbp2(aa):
[0196]
[0197]
car
‑
cd19第二代
‑
sbp3(nt):
[0198]
[0199]
car
‑
cd19第二代
‑
sbp3(aa):
[0200][0200][0201]
载体可以是表达载体。载体可是质粒载体。优选地,载体是慢病毒载体。
[0202]
在本发明范围内,慢病毒载体摂指非复制型载体,其供于使用包含顺式作用慢病毒rna或dna序列的转基因转导宿主细胞,且需要以反式形式提供的慢病毒蛋白(例如gag、pol和/或env)。慢病毒载体缺少功能性gag、pol和env蛋白的表达。慢病毒载体可以rna或dna分子的形式存在,这取决于所述逆转录载体的产生或发展阶段。
[0203]
慢病毒载体可以是重组dna分子的形式,如质粒。慢病毒载体可以是慢病毒载体颗粒的形式,如慢病毒和其他蛋白质的复合物中的rna分子。通常,对应于修饰或重组的慢病毒颗粒的慢病毒颗粒载体包含由两个单链rna拷贝组成的基因组。这些rna序列可通过从插入宿主细胞基因组的双链dna序列(原病毒载体dna)转录获得,或者可由转化的宿主细胞中质粒dna(质粒载体dna)的瞬时表达获得。
[0204]
优选地,慢病毒载体颗粒具有整合能力。因此,它们含有功能性整合酶蛋白。非整合载体颗粒有一个或多个突变,其消除了慢病毒载体颗粒的大部分或全部整合能力。例如,非整合载体颗粒可在由慢病毒pol基因编码的整合酶中含有突变,其导致整合能力降低。相反,整合载体颗粒包含功能性整合酶蛋白,其不含任何消除慢病毒载体颗粒的大部分或全部整合能力的突变。
[0205]
慢病毒载体来源于慢病毒,尤其是人免疫缺陷病毒(hiv
‑
1或hiv
‑
2)、猿免疫缺陷病毒(siv)、马传染性脑炎病毒(eiav)、山羊关节炎脑炎病毒(caev)、牛免疫缺陷病毒(biv)和猫免疫缺陷病毒(fiv),其经修饰以除去涉及致病性的遗传决定簇并导入对获得治疗效果有用的新决定簇。
[0206]
这类载体基于顺式和反式作用序列的分离。为制备复制缺陷型载体,可删除反式作用序列(如gag、pol、tat、rev和env基因)并使用编码转基因的表达盒代替。
[0207]
非分裂细胞中的有效整合与复制通常需要慢病毒基因组中心存在两种顺式作用序列:中心多嘌呤序列(cppt)和中心终止序列(cts)。这导致形成三链dna结构,称为中心dna“瓣(flap)”,其作为对核孔处的整合前复合物脱包被并将表达盒有效导入非分裂细胞(如树突细胞)的细胞核的信号。
[0208]
在一个实施方式中,本发明包括包含称为cppt/cts序列的中心多嘌呤序列和中心终止序列,具体地,在欧洲专利申请ep 2169073中。
[0209]
其它序列通常以顺式出现,如参与载体原病毒dna序列整合到宿主细胞基因组的长末端重复(ltr)。可通过使ltr序列突变以获得载体,例如在所述ltr的结构域u3中突变(δu3)(miyoshi h等,1998,j virol.72(10):8150
‑
7;zufferey等,1998,j virol 72(12):9873
‑
80)。
[0210]
优选地,载体不含增强子。在一个实施方式中,本发明包括包含ltr序列的慢病毒载体,优选具有移除了3’ltr中启动子和增强子序列的突变的u3区(δu3)。
[0211]
也可整合包装序列ψ(psi)以辅助多核苷酸序列衣壳化成载体颗粒(kessler等,2007,leukemia,21(9):1859
‑
74;paschen等,2004,cancer immunol immunother 12(6):196
‑
203)。
[0212]
在一个实施方式中,本发明包括包含慢病毒包装序列ψ(psi)的慢病毒载体。
[0213]
其它额外的功能性序列,如转运rna
‑
结合位点或引物结合位点(pbs)或土拨鼠后调控元件(wpre)也可优选包含在本发明的慢病毒载体多核苷酸序列中,以得到转基因在体内更稳定的表达。
[0214]
在一个实施方式中,本发明包括包含pbs的慢病毒载体。在一个实施方式中,本发明包括包含wpre和/或ires的慢病毒载体。
[0215]
因此,在优选的实施方式中,慢病毒载体包含至少一种cppt/cts序列、一种ψ序列、一种(优选2种)ltr序列和包括在β2m或mhc i型启动子转录控制下的转基因的表达盒。
[0216]
启动子
[0217]
本发明包括使用启动子来驱动在t细胞(优选人t细胞)中从慢病毒载体高表达car。优选的启动子是人泛素、mhc i型、mhc ii型、和β2微球蛋白(β2m)启动子。
[0218]
在多个实施方式中,启动子在抗原递呈细胞(包括树突细胞)中驱动高表达。优选地,该启动子缺少增强子元件以避免插入效应。
[0219]
最优选地,启动子不是cmv启动子/增强子。优选地,启动子不是dectin
‑
2或mhcii启动子。
[0220]
多种哺乳动物(人)mhc i型启动子的序列如下所示:
[0221]
hla
‑
a2(mhc i):
[0222][0223]
hla
‑
b7(mhc i):
[0224][0225]
hla
‑
cw5(mhc i):
[0226][0227]
hla
‑
e(mhc i):
[0228][0229]
hla
‑
f(mhc i):
[0230][0230]
[0231]
人β2
‑
微球蛋白启动子的序列如下所示:
[0232][0233]
人泛素(ubi)启动子的序列如下所示:
[0234][0235]
人hla
‑
drα启动子的序列如下所示:
[0236][0236][0237]
在多个实施方式中,慢病毒载体包含β2m、ubi、mhc ii或mhc i型启动子。优选地,mhc i型启动子是hla
‑
a2启动子、hla
‑
b7启动子、hla
‑
cw5启动子、hla
‑
f或hla
‑
e启动子。在多个实施方式中,启动子序列包括与seq id no:9、seq id no:10、seq id no:11、seq id no:12、seq id no:13、seq id no:14、seq id no:38、或seq id no:41的启动子序列具有超过90%、优选超过95%、更优选超过99%相同性的多核苷酸序列。
[0238]
在一些实施方式中,启动子在bdca+树突细胞中的表达是该启动子在骨骼肌细胞中表达的至少10、12、15、20、25、30、35、40、50、或60倍。
[0239]
在一个实施方式中,本发明包括包含慢病毒载体的慢病毒载体颗粒,该慢病毒载体颗粒包含指导微生物或肿瘤抗原表达的树突细胞
‑
特异性启动子的慢病毒载体,其中慢病毒载体颗粒在bdcm细胞中显示比hek293t细胞中更高的抗原表达。
[0240]
本发明包括含有不含增强子的启动子的慢病毒载体。
[0241]
本发明包括将mhc i型(mhci)、ubi、ef1α或β2微球蛋白启动子(β2m)启动子插入慢病毒载体中。本文所用的“mhc i型(mhci)启动子”或“β2微球蛋白启动子”或“mhc ii型(mhcii)”或“人泛素启动子”包括天然产生的或合成的mhc i型启动子或β2微球蛋白启动子或mhc ii型启动子或人泛素启动子。术语“mhc i型启动子”不包括β2m启动子。
[0242]
在一个实施方式中,包含启动子的慢病毒载体颗粒在bdcm细胞中显示出比在hek293t细胞中更高的表达。
[0243]
启动子可以是天然产生的启动子。天然产生的启动子的示例是人β2m、hla
‑
a2、hla
‑
b7、hla
‑
cw5、hla
‑
e、hla
‑
f、hla
‑
drα和泛素基因启动子。
[0244]
这些天然产生的mhci启动子一般从编码mhc i型蛋白的基因的启动子区中克隆或复制,后称为推定的编码,如基因组数据库中的蛋白质(根据:ncbi多核苷酸数据库http://www.ncbi.nlm.nih.gov/guide/dna
‑
rna)。β2m和mhc i型蛋白都进入主要组织相容性复合物(mhc)。
[0245]
这些基因编码的蛋白质存在于几乎所有细胞类型中。mhci蛋白一般存在于白细胞膜的表面处,其中它们与β2
‑
微球蛋白(β2m)结合。这些相关联蛋白质的作用是将内源性来源的肽递呈至cd8+t细胞。因此,其在抗原特异性免疫应答的产生过程中起核心作用。因为多年来mhc i型蛋白已得到了广泛研究和描述,所以其基因已被成熟表征并可使用序列比对工具(如blast法)良好地检测(altschul,s.f.等(1990).basic local alignment search tool.(局部比对检索基本工具)j.mol.biol.215(3):403
‑
410)。
[0246]
mhc i型启动子也共有在抗原递呈细胞(包括树突细胞)中被强激活的能力,以及,在大多数其它人体组织中降低强度的能力。
[0247]
本发明的启动子还可含有调控元件,如一个或多个sp1和ets结合位点。在一个优选实施方式中,mhc i型启动子含有2个sp1结合位点和1个ets结合位点。在其他实施方式中,ap1和/或ap2位点也包含在启动子中。
[0248]
优选的启动子是天然产生的人β2m、hla
‑
a2、hla
‑
b7、hla
‑
cw5、hla
‑
e和hla
‑
f启动子。
[0249]
启动子也可以是合成的。合成的启动子包括使用分子生物学技术组装启动子的个体组分合成的启动子或者使用分子生物学技术由天然产生的启动子衍生的启动子。
[0250]
在多个实施方式中,合成的启动子包含与β2m、ubi、mhc ii或mhc i型基因启动子(seq id no:9
‑
14、38、或41)的启动子序列具有超过90%、优选超过95%、更优选超过99%、或100%相同性的多核苷酸序列。
[0251]
mhc型基因的转录通常由2种主要调控元件介导:干扰素刺激应答元件(isre)和sxy模块(包括w/s、x1x2/位点α以及y/增强子b调控元件)。参见van den elsen,immunogenetics(1998)48:208
‑
211。
[0252]
这些调控启动子元件位于延伸自大约转录起始位点上游第
‑
220至
‑
95核苷酸的区域中。其介导mhc i型基因的组织特异性和细胞因子诱导的转录。
[0253]
mhc i型基因启动子的isre通常含有干扰素调节因子(irf)家族成员的结合位点。因此,i型mhc启动子的一个特性是结合干扰素调节因子(irf)家族成员。这可由,例如,凝胶迁移试验验证。
[0254]
另一种调控元件,增强子a(含有细胞核转录因子κb(nf
‑
κb)的结合位点)存在于大多数情况中。因此,mhc i型启动子的一个特性是结合细胞核转录因子κb(nf
‑
κb)。这可由,例如,凝胶迁移试验验证。
[0255]
除了isre,mhc i型启动子通常共有另一组保守的上游序列基序,由3种调控元件组成:s或w盒、x1/crex2盒或位点α、以及y盒或增强子b,它们一起被称为sxy模块。该sxy模块通常与含有调节因子x(rfx;由rfx5、rfxb/ank和rfxap组成)、camp反应元件结合蛋白(creb)/激活转录因子(atf)和作为增强体驱动这些基因反式激活的核因子y(nfy)的多蛋白复合物协同地结合。因此,mhc i型启动子的一个特性是结合这些因子。这可由,例如,凝胶迁移试验验证。
[0256]
相反地,mhc ii型启动子不显示增强子a或isre元件(van den elsen,p.j等,1998,immunogenetics.48:208
‑
221)。此外,如使用从裸淋巴细胞综合征(bls;一种由于rfx亚基之一或ciita中发生突变产生的重症联合免疫缺陷)患者建立的细胞系进行的研究所示,发现ii型mhc基因调控中的rfx和ciita是至关重要的。此外,缺少ciita或rfx亚基之一分别影响mhc增强体的功能和组装,导致缺少ii型mhc且i型mhc转录水平下降(van den elsen,p.j.等,2004,current opinion in immunology,16:67
‑
75)。
[0257]
在一个实施方式中,本发明包括一种方法,其包括将本发明的启动子,尤其是β2m、ubi、mhc ii型、或mhci型启动子,插入慢病毒载体中以引导本发明的car的表达。该方法还可包括插入本文中所述任意其他核酸元件,如dna瓣序列。
[0258]
分离的细胞
[0259]
本发明包括细胞,尤其是人免疫系统的细胞,其包含编码本发明的car的慢病毒载体颗粒和载体。优选地,所述细胞是t细胞,包括tαβ和tγδ细胞,或nk细胞。
[0260]
在一个实施方式中,所述细胞含有整合到细胞基因组中的载体。在一个实施方式中,所述细胞含有瞬时表达car的载体。在一个实施方式中,所述细胞产生编码car的慢病毒载体颗粒。
[0261]
在多种实施方式中,本发明包括包含编码car的载体或慢病毒载体颗粒的细胞系,细胞群,或细胞培养物。
[0262]
慢病毒载体颗粒
[0263]
本发明提供产生慢病毒载体颗粒的方法。慢病毒载体颗粒(或慢病毒颗粒载体)包括与病毒蛋白质相关联的慢病毒载体。该载体优选是整合载体。
[0264]
在一个实施方式中,慢病毒载体颗粒编码本发明的car。
[0265]
在一个实施方式中,慢病毒载体颗粒包含hiv
‑
1 gag和pol蛋白。优选地,慢病毒载体颗粒包含d亚型,尤其是hiv
‑1ndk
,gag和pol蛋白。
[0266]
根据该方法的一个实施方式,慢病毒载体颗粒在转化有dna质粒的宿主细胞中获得。
[0267]
这种dna质粒可包含:
[0268]
‑
细菌复制起点(如puc ori);
[0269]
‑
用于选择的抗生素抗性基因(如kanr);且更具体地:
[0270]
‑
慢病毒载体,其包含至少一个编码通过转录连接至β2m、ubi、mhc ii型、或mhc i型启动子的car的核酸。
[0271]
该方法允许产生根据本发明的重组载体颗粒,包括以下步骤:
[0272]
i)使用慢病毒载体转染合适的宿主细胞;
[0273]
ii)使用包装质粒载体转染所述宿主细胞,所述包装质粒载体含有编码反转录病毒(优选慢病毒)的至少结构和聚合酶(+/
‑
整合酶)活性的病毒dna序列,;这类包装质粒在本领域中已有描述(dull等,1998,j virol,72(11):8463
‑
71;zufferey等,1998,j virol 72(12):9873
‑
80)。
[0274]
iii)培养所述转染的宿主细胞以使所述慢病毒载体表达并包装成慢病毒载体颗粒;以及
[0275]
iv)收获所述培养的宿主细胞中由步骤iii)的表达和包装所得到的慢病毒载体颗粒。
[0276]
基于不同原因,制备获得的逆转录病毒颗粒的假型(即增加或代替特定颗粒包膜蛋白)可能是有帮助的。例如,具有不同的包膜蛋白以区分重组颗粒与天然颗粒或其他重组颗粒可能是有利的。在疫苗接种策略中,当后者已经发展出针对慢病毒的免疫时,假型颗粒载体更有可能逃避免疫系统。如果类似的颗粒载体的连续注射是使患者对疾病产生免疫所必需的,那么这是特别有帮助的。
[0277]
为制备本发明的逆转录病毒颗粒的假型,还可使用编码病毒包膜蛋白(优选vsv
‑
g包膜蛋白)的一种或数种包膜dna质粒来转染宿主细胞。
[0278]
合适的宿主细胞优选是人培养细胞系,例如hek细胞系。
[0279]
或者用于生产载体颗粒的方法可在宿主细胞中进行,其基因组已使用如下一种或多种组分稳定转化:慢病毒载体dna序列、包装基因和包膜基因。这类dna序列可被视作与本发明的原病毒载体类似,包含额外的启动子以允许载体序列转录并提高颗粒生产率。
[0280]
在优选实施方式中,还对宿主细胞进行修饰,使其能够在培养基中以连续的方式生产病毒颗粒,同时整个细胞不发生溶胀或死亡。关于用于生产病毒颗粒的这类技术,可参见strang等,2005,j virol 79(3):1165
‑
71;relander等,2005,molther 11(3):452
‑
9;stewart等,2009,gene ther,16(6):805
‑
14;和stuart等,2011,hum gene ther。
[0281]
本发明的目的由用慢病毒颗粒载体转化的宿主细胞组成。
[0282]
慢病毒颗粒载体可包括以下元件,如前文所定义:
[0283]
‑
cppt/cts多核苷酸序列;以及
[0284]
‑
编码β2m、ubi或mhc i型启动子控制下的car的核酸,和可选的上文所述其他元件中的一种。
[0285]
优选地,慢病毒载体颗粒的剂量是106、2x106、5x106、107、2x107、5x107、108、2x108、5x108、或109tu。
[0286]
细胞中表达car的方法
[0287]
本发明包括在细胞,尤其是t细胞,并且优选增殖的t细胞中表达car的方法。该方
法包括在允许car表达的条件下,使用本发明的慢病毒载体或慢病毒颗粒载体转导细胞,并优选增殖该t细胞。
[0288]
细胞优选是哺乳动物细胞,特别是人细胞。特别优选的是人非分裂细胞。
[0289]
优选地,细胞是原代t细胞或nk细胞。
[0290]
该方法还可包括收获或分离car。
[0291]
慢病毒载体或慢病毒颗粒载体优选包含本发明的启动子。
[0292]
在一个实施方式中,本发明包括用生物素处理细胞以从hook释放car。优选地,用至少0.2、0.4、0.8、1.6、2.5、5、10、20、40、或80μm的初始浓度的生物素处理细胞。
[0293]
在一个实施方式中,本发明包括表达car的方法,包括向慢病毒载体中插入β2m、ubi、或mhci启动子,使其指导编码car的核酸的表达并用含有该启动子的载体转导细胞,优选t或nk细胞,并任选用至少0.2、0.4、0.8、1.6、2.5、5、10、20、40、或80μm的初始浓度的生物素处理细胞。
[0294]
慢病毒载体的治疗性用途
[0295]
本发明还涉及本发明的慢病毒载体,尤其是慢病毒载体颗粒形式,用于制备治疗性组合物或疫苗的应用,所述治疗性组合物或疫苗能够诱导或造成与载体编码的car的免疫反应发生或该反应的增强。
[0296]
本发明包括向人给予慢病毒的载体(或“慢病毒载体”)的方法。优选地,慢病毒颗粒是包含功能性整合酶蛋白的整合的慢病毒颗粒。
[0297]
优选的给药模式包括再灌注修饰的t细胞,优选静脉内或关节内给药,最优选肿瘤内给药。
[0298]
在一个实施方式中,本发明包括在人中诱导免疫应答的方法,包括向t或nk细胞给予包含功能性整合酶蛋白的慢病毒载体合理和慢病毒载体并向人给予该修饰的细胞;其中整合慢病毒载体包括引导car表达的启动子;并产生与car的免疫反应。
[0299]
本发明还可用于针对肿瘤和癌症的治疗方案,特别可用于针对癌症和肿瘤的免疫治疗或疫苗接种治疗的方案。
[0300]
本发明还涉及包含前文所定义的慢病毒载体的免疫原性组合物。
[0301]
本发明的免疫原性组合物优选包含载体和载体颗粒中的cppt和cts序列,以诱导或刺激载体基因组在靶细胞中的核输入。
[0302]
逆转录期间,cppt和cts序列诱导称作dna三链体的三链dna结构的形成,其刺激dna载体序列的核输入。优选地,载体包含car以及逆转录病毒或逆转录病毒样来源逆转录、表达和壳体化的调控信号。
[0303]
本发明的慢病毒载体具有重定向t淋巴细胞和/或其他免疫细胞的特异性和功能的能力。它们可快速生成靶向特异性肿瘤抗原或与其他病变如自免疫疾病相关的抗原的t细胞。
[0304]
本发明的慢病毒载体可用于治疗方法和诱导免疫应答的方法,包括向细胞,优选t或nk细胞给予慢病毒载体,向宿主给予细胞,并产生重定向t淋巴细胞和/或其他免疫细胞的特异性和功能的特异性免疫应答。
[0305]
本发明的免疫原性组合物的特定优势在于,它们可用于重定向t淋巴细胞和其他免疫细胞针对多种抗原的特异性和功能,载体或载体颗粒中的car指向这些抗原。
[0306]
因此,本发明包括可用于治疗性免疫接种方案的组合物。
[0307]
具体而言,其可与佐剂、其他免疫原性组合物、化疗或任何其他治疗性处理联用。
[0308]
本发明包括用于向人给药的包含慢病毒载体颗粒的组合物,该慢病毒载体颗粒包含功能性整合酶蛋白和慢病毒载体;其中慢病毒载体的dna包含引导包含或由car组成的氨基酸表达的启动子。
[0309]
在一个实施方式中,本发明包括向人给予,优选通过肌肉内给予编码嵌合抗原受体的慢病毒载体、或由该慢病毒载体转导的细胞,该嵌合抗原受体包含结合结构域;跨膜结构域;hook
‑
结合结构域,优选包含链霉亲和素
‑
结合肽;和活化结构域,包含seq id no:3、seq id no:4、seq id no:5、seq id no:6、seq id no:7、或seq id no:8、seq id no:33、或seq id no:34的至少100个氨基酸的t细胞活化片段。优选地,慢病毒载体还包含hook,优选包含链霉亲和素蛋白,最优选包含seq id no:32、seq id no:33或其突变体的氨基酸序列,该突变体在氨基酸49处具有甘氨酸突变,优选突变成苏氨酸。优选地,hook
‑
结合结构域包含seq id no:1的氨基酸序列或由seq id no:2的核酸序列编码。
[0310]
该方法还可包括向人给予生物素以从er或高尔基体释放car。优选地,给予至少0.2、0.4、0.8、1.6、3.2、5、10、20、40、或80μm的初始浓度的生物素。
[0311]
因此,已经描述了本发明的不同实施方式,本领域技术人员应注意,本文所公开的内容仅是示例性的,且多种其他替代、调整或修改可包括在本发明的范围内。因此,本发明不限于本文所示特定实施方式。
实施例
[0312]
实施例1.分子构建
[0313]
使用分别包括sdei和xbai的限制性位点的正向(5
’‑
cttactagttggaagggctaattcactcccaac
‑3’
;seq id no:15)和反向(5
’‑
cattctagaactgctagagattttccacactg
‑3’
;seq id no:16)寡核苷酸进行对ptripδu3
‑
cmv
‑
gfp(15)的原病毒区的pcr扩增。所得片段在pvax
‑
1质粒(英杰公司,生命技术公司)的spei和xbai位点之间消化并克隆,从中已经删除了mlui位点。产生的质粒被命名为pflap
‑
cmv
‑
gfp。sv40序列通过pcr从ptripδu3
‑
cmv
‑
gfp质粒扩增(使用5
’‑
taccccgggccatggcctccaaaaaagcctcctcactacttc
‑3’
(seq id no:17)和5
’‑
actcccgggtaattttttttatttatgcagaggccgaggccgcc
‑3’
(seq id no:18)寡核苷酸),并且克隆到pflap
‑
cmv
‑
gfp的pml1位点中,然后将所得质粒命名为pflap
‑
cmv
‑
gfp
‑
sv。用分别包括mlui和bamhi位点的正向(5
’‑
tacacgcgtggagttccgcgttacataacttacgg
‑3’
;seq id no:19)和反向(5
’‑
cgtggatccgatcgcggtgtcttctatggaggtcaaaac
‑3’
;seq id no:20)寡核苷酸来扩增cmv启动子。所得的片段被克隆回pflap
‑
cmv
‑
gfp
‑
sv的mlui和bamhi位点之间,使得易于替换慢病毒载体内的启动子。然后通过pcr用针对β2m启动子的5
’‑
gccggcgcgccgagaaaccctgcagggaattccc
‑3’
(seq id no:21)和5
’‑
cgtggatccgatcgctcggcccgaatgctgtcagcttcagg
‑3’
(seq id no:22)从hek 293t细胞dna扩增启动子,并且克隆到pflap
‑
cmv
‑
gfp
‑
sv的mlui和bamh1位点之间以产生pflap
‑
β2m
‑
sv。扩增的β2m启动子序列如下:
[0314][0314]
car可在pflap
‑
β2m
‑
sv的bamhi和xhoi位点之间,在gfp基因的位置上合成并克隆。
[0315]
例如,可由bamhi和xhoi消化pflap
‑
β2m
‑
gfp
‑
sv并且可在这些位点之间,在gfp基因的位置上可令含多个克隆位点(mcs,携带sali,sacii,ndei,asci和nhei限制性位点)的dna接头以允许插入编码car的核酸序列。
[0316]
通过pcr扩增hiv
‑
1ndk基因组来构建包装质粒pthv
‑
gp
‑
n(使用以下寡核苷酸,5
’‑
atgcatgcgtcgacctcgagttaatcctcatcctgtctacttgccac
‑3’
(seq id no:24)和5
’‑
gcatgcatcggccggggcggcgactggtgagaggccaccatgggtgcgagagcgtcagtattaag
‑3’
(seq id no:25))。所得的片段用eagi和sali限制性酶消化并插入p8.74包装质粒(15)中,已在之前从该质粒中去除了eag1
‑
sali片段。
[0317]
通过合成对应于水泡性口炎病毒indiana(genbank#cax62728.1)、new jersey(genbank#cax62729.1)和cocal(genbank#cax62731.1)毒株的密码子优化基因来生成假型化质粒。然后,这些基因用ecor1和bamh1消化并且克隆到pvax1质粒(英杰公司,生命技术公司)的相应限制性位点之间。
[0318]
可按照生产商的说明(mn公司)使用nucleobond xtra maxi ef柱来产生质粒。
[0319]
实施例2.慢病毒产生
[0320]
r&d产生:可如之前所述通过hek 293t的瞬时磷酸钙转染来产生载体(25)。将hek 293t(人胚胎肾细胞系,atcc crl
‑
11268,(graham等,1977))细胞保持在补充了10%胎牛血清(fbs,paa)、1%l
‑
谷氨酰胺(eurobio)、1%青霉素
‑
链霉素(生命技术公司(life technologies)的gibco)和1%丙酮酸钠(生命技术公司的gibco)的达氏改良伊氏培养基(dmem/高改良,hyclone)中。该细胞系保持在37℃下含5%co2的湿润气氛的孵育器中。使用标准磷酸钙沉淀方案通过hek 293t细胞的瞬时转染产生慢病毒载体。hek 293t细胞以7x106个细胞接种在10ml完全培养基中的10cm2组织培养皿(bd falcon)中并在37℃下5%co2的潮湿气氛的孵育器中保持24小时以粘附。对于各产生的载体,如下转染组织培养皿:慢病毒主链质粒pflap
‑
启动子
‑
car_cd19(10μg)、pthv
‑
env1编码包膜质粒(2μg)和pthv
‑
gp
包装质粒(10μg)与353μl的无菌蒸馏水(生命技术公司的gibco)和125μl的cacl2(伏路卡公司(fluka))混合。然后向500μl的37℃预热的hbs 2x ph=7.3中逐滴加入dna混合物,并且1ml的所得沉淀物加入细胞的培养基中。转染的细胞然后在37℃,5%co2下孵育。培养基在转染后24小时用7ml不含血清的收获培养基置换并且在另外24小时后通过2500rpm下离心5分钟收获病毒上清。然后,收获的澄清体培养物(210ml)在mgcl2(西格玛奥德里奇公司)存在下用dna酶(罗氏公司)处理30分钟以避免残留转染dna,并且通过在4℃,22000rpm下离心1小时超浓缩。各载体团块在70μl pbs
‑
乳糖(40mg/l)中重悬,汇集,30μl等分并储存在
‑
70℃
±
10℃下。
[0321]
对于产物表征和药物释放,可按照以下进行质量测试:疫苗的法规文本:疫苗所要求的质量控制按照欧洲药典(6.16部分),“活重组病毒载体疫苗的质量、非临床和临床方面指南”(ema/chmp/141697/2009),“慢病毒载体的开发和生产指南”(chmp/bwp/2458/03);基因治疗药物产物的法规文本:人类使用的转基因药物产物所要求的质量控制按照欧洲药典(5.14部分),基因治疗产物特定的质量控制,如“转基因移走无产物的质量、临床前和临床方面的指南注意”(chmp/bwp/3088/99)中所定义;对生物技术产品的法规文本(ich q5a至ich q5e);对说明书的法规文本(ich q6a和ich q6b)以及按照欧洲药典(7.0部分)的胃肠外制剂所需的质量控制。
[0322]
实施例3.慢病毒载体效价
[0323]
qpcr反应:hek 293t细胞接种在培养基的6
‑
孔平板(bd falcon)并在37℃,5%co2下的潮湿气氛中孵育4小时。用慢病毒载体的3个连续稀释来转导细胞。孵育后72小时,细胞收获并且产生经转导的hek 293t细胞团块。使用基于qiagen qiaamp dna小试剂盒手册的方法从转导的细胞团块中提取全基因组dna。使用taqman qpcr来进行原病毒定量。用主混合物(富酶泰斯热科学公司(fermentas thermo scientific))、flap a引物(cccaagaacccaaggaaca;seq id no:26)和flap s引物(agacaa gatagaggaagagcaaaac;seq id no:27)引物、和lenti tm探针(6fam
‑
aaccattaggagtagcacccaccaagg
‑
bbq;seq id no:52)来进行扩增。用肌动蛋白基因(相同混合物,肌动蛋白a
‑
cggtgaggatcttcatgaggtagt
‑
(seq id no:28),肌动蛋白s
‑
aacaccccagccatgtacgt
‑
(seq id no:29)引物和humura act tm探针
‑
6fam
‑
ccagccaggtccagacgcagga
‑
bbq
‑
(seq id no:30)的定量来进行标准化。在mastercycler ep realplex s(eppendorf,50℃下2分钟,95℃下10分钟,和40个循环的95℃下15秒和63℃下1分钟)上进行两种反应。在mastercycler ep realplex软件上进行分析。
[0324]
实施例4.调节的car
[0325]
生成编码car的慢病毒载体。car_cd19第二代和第三代序列(seq id no:39和seq id no:40)购自geneart(生命技术公司),并克隆替换pflap
‑
δu3
‑
β2m
‑
gfp、pflap
‑
δu3
‑
hla
‑
a2
‑
gfp、pflap
‑
δu3
‑
hla
‑
drα
‑
gfp或pflap
‑
δu3
‑
ubc
‑
gfp的bamhi和xhoi限制性位点之间的gfp基因,取决于所需的启动子。
[0326]
car在3个不同位置上以与sbp的融合蛋白生成。慢病毒载体经进一步修饰以含有与核心链霉亲和素蛋白融合的er的hook。启动子是β2m启动子。
[0327]
这些慢病毒载体将被用于癌症的体外和动物模型。将以40μm(较高和较低浓度的滴定)的初始浓度给予生物素以测试从er的释放。首先,将体外分析car迁移到细胞表面(在
结合结构域位置上或信号序列和结合结构域之间的gfp)。接着,通过注射car
‑
t细胞和评价在来自动物的细胞表面上的迁移(gfp)在动物模型(小鼠和大鼠)进行同样分析。
[0328]
将同时生成含有hook
‑
结合结构域(sbp)的“第二代”car(2个内胞质活化结构域)和“第三代”car(3个内胞质活化结构域)。初始时,评价的结合结构域将是抗
‑
cd19、抗
‑
pdl
‑
1、抗
‑
pd1、抗
‑
hedgehog、抗
‑
cd123、和抗
‑
cd123/cd33。
[0329]
carmin 1.0:针对cd19(cd19+白血病和淋巴瘤)、lmp
‑
1和
‑
2(ebv
‑
诱导的白血病)的第二代(含cd3_和4
‑
1bb共信号转导结构域)和第三代(含cd3_、cd28和4
‑
1bb结构域)car的慢病毒载体。慢病毒载体允许t细胞中car的优化表达,并且进行对car
‑
t细胞的功效的影响的研究。血液恶性肿瘤可用作基准。
[0330]
carmin 2.0;开发可切换开/关系统,其基于通过hook锚定在内质网(er)的膜的w蛋白,及其被导入car结构中的结合伴侣y。x
‑
hook(例如,链霉亲和素)和y(例如,链霉亲和素
‑
结合蛋白)
‑
car之间的相互作用使得car在er内滞留。z蛋白(例如,生物素)的添加替代了x向z而不是y的结合的平衡,由此导致car从er中释放并且其表达到胞质膜中。将通过z消耗(或拮抗剂)停止car的释放,并且可易于通过z诱导物的再导入来再活化其余细胞。
[0331]
hook和car可载体化于一个慢病毒载体中并且可用于临床中(b2m
‑
hook
‑
ires
‑
car)。可在固定的细胞(hek293t、jurkat、hela)上和在原代细胞(t
‑
细胞)上体外进行评价。该系统将增加car
‑
t细胞的安全性。可切换cd19
‑
car系统可体外评价表达并体内评价功效。
[0332]
大部分使用至今的scfv是鼠源的。针对这些鼠scfv的中和抗体可显示car的功效。或者,我们将开发驼科纳米抗体以用作结合结构域,因为它们与人抗体vh结构域高度同源并且它们显示高抗原结合能力。将用含有针对her2的纳米抗体作为结合结构域的第二代car来进行概念验证。这些技术平台允许car设计、产生和评价中的弹性和反应性,因此生成优化car
‑
t细胞。这种差异诱导和可逆的(开/关)car t
‑
细胞技术旨在患者床边进行递送(自动化过程)。
[0333]
实施例5.人t细胞中的表达
[0334]
通过在ficoll上的梯度密度离心从外周血纯化外周血单核细胞(pbmc)。在pbmc洗涤之后,通过使用泛t细胞分离试剂盒(美天旎公司)进行负磁性选择(即,不需要的细胞经磁性标记而t细胞保持不接触)来纯化cd3+t细胞。需要该步骤以获得高度纯化的t细胞群。根据在该分离步骤后获得的产率,107至108个t细胞在2,5x106/ml下在优化无血清细胞培养基中培养,该培养基开发用于人t细胞的培养和增殖(texmacs培养基,美天旎公司)。这些t细胞通过来自美天旎公司的t细胞活化/增殖来活化。试剂盒由抗
‑
生物素macsibead颗粒以及针对人cd2、cd3和cd28的生物素化的抗体组成。加载生物素化抗体的抗
‑
生物素macsibead颗粒用于模拟抗原呈递细胞并活化t细胞。通过每2个细胞使用1个加载的抗生物素macsibead颗粒来完成t细胞的最优活化。t细胞被活化3天。然后在4的moi下进行活化的t细胞的转导,其表示用4个转导单位的慢病毒颗粒来孵育1个t细胞。通过流式细胞术评价48或72小时慢病毒颗粒添加的car表达,通过用生物素化的山羊抗
‑
小鼠igg,之后用与藻红蛋白偶联的链霉亲和素对鼠cd19
‑
结合结构域染色。由cd3、cd4和cd8染色来表征t细胞亚群。这使得能够在t细胞的表面上特异性检测到car。整个过程用texmacs培养基进行,允许t细胞的存活和增殖。
[0335]
实施例6.car
‑
rush的表达和结构
[0336]
从国家血液服务机构(法国伦吉斯)获得合格血液。通过ficoll(淋巴细胞分离培养基,eurobio)梯度密度分离来纯化外周血单核细胞(pbmc)。然后通过使用泛t细胞分离试剂盒(美天旎公司)的磁性分离来从pbmc纯化t细胞。按照生产商的指令分离t细胞。在37℃/5%co2下在texmacs培养基(美天旎公司)中以2,5x106个细胞/ml的浓度将细胞置于培养基中,并通过来自美天旎公司的t细胞活化/增殖试剂盒(按照生产商的说明制备的抗
‑
cd2/
‑
cd3/
‑
cd28纳米颗粒)以1∶2的珠∶t细胞比率活化3天。活化后,收获t细胞,经计数并在37℃/5%co2下置于24孔板的texmacs培养基中培养。通过向孔中直接添加不同moi的慢病毒载体来进行转导。测试的不同慢病毒载体是:(i)含有4
‑
1bb和cd3ζ胞内结构域的第二代抗
‑
cd19 car;(ii)在3个不同位置上含有链霉亲和素结合蛋白的相同载体(car
‑
sbp1,car
‑
sbp2,car
‑
sbp3)。
[0337]
如下计算按照moi加入各孔的载体的体积:待加入体积(ul)=(moi x细胞数(以百万计)/载体浓度(转导单位/ml))x1000。
[0338]
用3.109tu/ml的载体效价以10的moi转导200000个细胞
‑
>待加入载体体积(μl)=(10x0.2x106/3x109)x1000=0.51μl。在转导那天以50iu/ml加入人重组il
‑
2(美天旎公司)。在第3天,收获转导的t细胞,用dpbs1x充分洗涤并且在96孔板中进行对感兴趣分子的免疫染色。t细胞首先用与efluor 780偶联的活力染料(可固定活力染料,电子生物科学公司(ebiosciences))孵育,并在4℃下孵育30分钟。孵育在无叠氮化物和无蛋白质的dpbs1x中进行。细胞然后用dpbs 1x洗涤并且用生物素化的山羊抗
‑
小鼠igg(fab’)2(杰克逊免疫研究公司)在4℃下孵育30分钟。孵育后,细胞用automacs运行缓冲剂(美天旎公司)洗涤并且使用与藻红蛋白(杰克逊免疫研究公司)偶联的链霉亲和素和与pe
‑
cy7(bd生物科学公司)偶联的小鼠抗
‑
人cd3的混合物来进行第三次孵育。孵育在4℃下进行30分钟。在第三次孵育之后,细胞在automacs运行缓冲剂中洗涤,并在macsquant分析仪(美天旎公司)上进行数据采集之前在cellfix(bd生物科学公司)中固定。使用flowjo软件来分析流式细胞数据。
[0339]
实施例7.在与编码hook
‑
链霉亲和素的慢病毒载体共转导和生物素处理之后car
‑
rush构建体的表达和表现。
[0340]
从国家血液服务机构(法国伦吉斯)获得合格血液。通过ficoll(淋巴细胞分离培养基,eurobio)梯度密度分离来纯化外周血单核细胞(pbmc)。然后通过使用泛t细胞分离试剂盒(美天旎公司)的磁性分离来从pbmc纯化t细胞。按照生产商的指令分离t细胞。在37℃/5%co2下在texmacs培养基(美天旎公司)中以2,5x106个细胞/ml的浓度将细胞置于培养基中,并通过来自美天旎公司的t细胞活化/增殖试剂盒(按照生产商的说明制备的抗
‑
cd2/
‑
cd3/
‑
cd28纳米颗粒)以1∶2的珠∶t细胞比率活化3天。
[0341]
活化后,收获t细胞,经计数并在37℃/5%co2下置于24孔板的texmacs培养基中培养。通过向孔中直接添加不同moi的慢病毒载体来进行共转导。测试的2个慢病毒载体是:(i)hook
‑
链霉亲和素载体和(ii)在3个不同位置上含有链霉亲和素结合蛋白的4
‑
1bb和cd3ζ胞内结构域的第二代抗
‑
cd19 car(car
‑
sbp1,car
‑
sbp2和car
‑
sbp3)。
[0342]
如下计算按照moi加入各孔的各载体的体积:待加入体积(μl)=(moi x细胞数(以百万计)/载体浓度(转导单位/ml))x1000。
[0343]
在转导当天加入人重组il
‑
2(50iu/ml;美天旎公司)和生物素(40μm;西格玛
‑
奥德里奇公司)。
[0344]
在第3天,收获转导的t细胞,用dpbs 1x充分洗涤并且在96孔板中进行对感兴趣分子的免疫染色。t细胞首先用与efluor 780偶联的活力染料(可固定活力染料,电子生物科学公司(ebiosciences))孵育,并在4℃下孵育30分钟。孵育在无叠氮化物和无蛋白质的dpbs1x中进行。细胞然后用dpbs 1x洗涤并且用生物素化的山羊抗
‑
小鼠igg(fab’)2(杰克逊免疫研究公司)在4℃下孵育30分钟。孵育后,细胞用automacs运行缓冲剂(美天旎公司)洗涤并且使用与藻红蛋白(杰克逊免疫研究公司)偶联的链霉亲和素和与pe
‑
cy7(bd生物科学公司)偶联的小鼠抗
‑
人cd3的混合物来进行第三次孵育。孵育在4℃下进行30分钟。在第三次孵育之后,细胞在automacs运行缓冲剂中洗涤,并在macsquant分析仪(美天旎公司)上进行数据采集之前在cellfix(bd生物科学公司)中固定。使用flowjo软件来分析流式细胞数据。
[0345]
实施例8.car
‑
rush双顺反子构建体的表达和表现
[0346]
从国家血液服务机构(法国伦吉斯)获得合格血液。通过ficoll(淋巴细胞分离培养基,eurobio)梯度密度分离来纯化外周血单核细胞(pbmc)。然后通过使用泛t细胞分离试剂盒(美天旎公司)的磁性分离来从pbmc纯化t细胞。按照生产商的指令分离t细胞。在37℃/5%co2下在texmacs培养基(美天旎公司)中以2,5x106个细胞/ml的浓度将细胞置于培养基中,并通过来自美天旎公司的t细胞活化/增殖试剂盒(按照生产商的说明制备的抗
‑
cd2/
‑
cd3/
‑
cd28纳米颗粒)以1∶2的珠∶t细胞比率活化3天。
[0347]
活化后,收获t细胞,经计数并在37℃/5%co2下置于24孔板的texmacs培养基中培养。通过向孔中直接添加不同moi的慢病毒载体来进行转导。测试的不同慢病毒载体是:(i)“经典”第二代抗
‑
cd19 car;(ii)含有hook
‑
链霉亲和素、ires和第二代抗
‑
cd19 car的三种构建体,该第二代抗
‑
cd19 car含有4
‑
1bb和cd3ζ胞内结构域,具有在三个不同位置的链霉亲和素结合蛋白(hook
‑
ires
‑
car
‑
sbp1,hook
‑
ires
‑
car
‑
sbp2,hook
‑
ires
‑
car
‑
sbp3)。
[0348]
如下计算按照moi加入各孔的载体的体积:待加入体积(ul)=(moi x细胞数(以百万计)/载体浓度(转导单位/ml))x1000。
[0349]
测试的不同moi是10、20或30,取决于实验。
[0350]
在转导当天加入人重组il
‑
2(50iu/ml;美天旎公司)和生物素(40μm;西格玛
‑
奥德里奇公司)。
[0351]
在第3天和第7天,收获转导的t细胞,用dpbs 1x充分洗涤并且在96孔板中进行对感兴趣分子的免疫染色。t细胞首先用与efluor 780偶联的活力染料(可固定活力染料,电子生物科学公司(ebiosciences))孵育,并在4℃下孵育30分钟。孵育在无叠氮化物和无蛋白质的dpbs1x中进行。细胞然后用dpbs 1x洗涤并且用生物素化的山羊抗
‑
小鼠igg(fab’)2(杰克逊免疫研究公司)在4℃下孵育30分钟。孵育后,细胞用automacs运行缓冲剂(美天旎公司)洗涤并且使用与藻红蛋白(杰克逊免疫研究公司)偶联的链霉亲和素和与pe
‑
cy7(bd生物科学公司)偶联的小鼠抗
‑
人cd3的混合物来进行第三次孵育。孵育在4℃下进行30分钟。在第三次孵育之后,细胞在automacs运行缓冲剂中洗涤,并在macsquant分析仪(美天旎公司)上进行数据采集之前在cellfix(bd生物科学公司)中固定。使用flowjo软件来分析流式细胞数据。
[0352]
实施例9.car
‑
rush系统切换评价(关/开/关)
[0353]
从国家血液服务机构(法国伦吉斯)获得合格血液。通过ficoll(淋巴细胞分离培
养基,eurobio)梯度密度分离来纯化外周血单核细胞(pbmc)。然后通过使用泛t细胞分离试剂盒(美天旎公司)的磁性分离来从pbmc纯化t细胞。按照生产商的指令分离t细胞。在37℃/5%co2下在texmacs培养基(美天旎公司)中以2,5x106个细胞/ml的浓度将细胞置于培养基中,并通过来自美天旎公司的t细胞活化/增殖试剂盒(按照生产商的说明制备的抗
‑
cd2/
‑
cd3/
‑
cd28纳米颗粒)以1∶2的珠∶t细胞比率活化3天。
[0354]
活化后,收获t细胞,经计数并在37℃/5%co2下置于24孔板的texmacs培养基中培养。通过向孔中直接添加不同moi的慢病毒载体来进行转导。测试的不同慢病毒载体是:(i)“经典”第二代抗
‑
cd19 car;(ii)含有hook
‑
链霉亲和素、ires和第二代抗
‑
cd19 car的三种构建体,该第二代抗
‑
cd19 car含有4
‑
1bb和cd3ζ胞内结构域,具有在三个不同位置的链霉亲和素结合蛋白(hook
‑
ires
‑
car
‑
sbp1,hook
‑
ires
‑
car
‑
sbp2,hook
‑
ires
‑
car
‑
sbp3)。
[0355]
如下计算按照moi加入各孔的载体的体积:待加入体积(μl)=(moix细胞数(以百万计)/载体浓度(转导单位/ml))x1000。
[0356]
测试的不同moi是10和20。
[0357]
在转导那天加入人重组il
‑
2(50iu/ml;美天旎公司)和生物素(40μm;西格玛
‑
奥德里奇公司)。
[0358]
在第3天,细胞经洗涤并且以40μm添加生物素或不添加以再诱导car
‑
sbp表达。
[0359]
在第7天,收获转导的t细胞,用dpbs 1x充分洗涤并且在96孔板中进行对感兴趣分子的免疫染色。t细胞首先用与efluor 780偶联的活力染料(可固定活力染料,电子生物科学公司(ebiosciences))孵育,并在4℃下孵育30分钟。孵育在无叠氮化物和无蛋白质的dpbs1x中进行。细胞然后用dpbs 1x洗涤并且用生物素化的山羊抗
‑
小鼠igg(fab’)2(杰克逊免疫研究公司)在4℃下孵育30分钟。孵育后,细胞用automacs运行缓冲剂(美天旎公司)洗涤并且使用与藻红蛋白(杰克逊免疫研究公司)偶联的链霉亲和素和与pe
‑
cy7(bd生物科学公司)偶联的小鼠抗
‑
人cd3的混合物来进行第三次孵育。孵育在4℃下进行30分钟。在第三次孵育之后,细胞在automacs运行缓冲剂中洗涤,并在macsquant分析仪(美天旎公司)上进行数据采集之前在cellfix(bd生物科学公司)中固定。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1