麦长管蚜E-β-法尼烯溶液处理下稳定表达的内参基因RPL11序列、克隆方法及应用
麦长管蚜e
‑
β
‑
法尼烯溶液处理下稳定表达的内参基因rpl11序列、克隆方法及应用
1.本技术为申请号201910608195.0(发明名称:麦长管蚜内参基因的部分序列、克隆方法及应用)的分案申请。
技术领域
2.本发明属于分子生物学技术领域,尤其涉及麦长管蚜内参基因sa
‑
hel基因、 sa
‑
rpl14基因、sa
‑
sa
‑
rpl11基因或sa
‑
28s基因部分序列、克隆方法及其作为内参基因在rt
‑
qpcr中的应用。
背景技术:
3.麦长管蚜(sitobion avenae fabricius)属同翅目,蚜科,是我国麦类作物的主要害虫之一,其寄主种类较多,除为害小麦、大麦、燕麦等麦类作物外,也为害水稻、高粱、玉米、甘蔗等其他禾本科作物以及早熟禾、看麦娘、马唐、棒头草、狗尾草、白羊草等禾本科、莎草科杂草。具有分布广、数量大、繁殖力强的特点,影响小麦植株的正常生长,导致小麦产量严重下降,给农业生产造成巨大损失。一般年份可使小麦减产5.1%~ 16.5%,大发生年份小麦减产40%以上。一般蚜虫都是无翅的,可受外界环境条件和生物因素调节生成有翅蚜和无翅蚜,有翅型个体可远距离迁飞寻找寄主植物,除直接刺吸小麦汁液外,还是传播麦类病毒病的重要媒介。
4.麦长管蚜无翅孤雌蚜体长3.1mm,宽1.4mm,长卵形,草绿色至橙红色,头部略显灰色,腹侧具灰绿色斑。触角、喙端节、跗节、腹管黑色,尾片色浅。腹部第六至八节及腹面具横网纹,无缘瘤。中胸腹岔短柄。额瘤显著外倾。触角细长,全长不及体长,第三节基部具1~4个次生感觉圈。喙粗大,超过中足基节,端节圆锥形,是基宽的1.8 倍。腹管长圆筒形,长为体长的1/4,在端部有十几行网纹。尾片长圆锥形,长为腹管的 1/2,有6~8根曲毛。有翅孤雌蚜体长3.0mm,椭圆形,绿色,触角黑色,第三节有8~ 12个感觉圈排成1行。喙不达中足基节。腹管长圆筒形,黑色,端部具15~16行横行网纹。前翅中脉三叉,分叉大。
5.在昆虫基因转录水平的研究中,实时荧光定量pcr技术(qrt
‑
pcr)是一种检测特定基因mrna表达水平和转录分析的有效手段。qrt
‑
pcr技术具有实时检测、特异性强、快速灵敏、准确定量的优点,实现了传统pcr从定性到精准定量的飞跃。进行qrt
‑
pcr 试验时,内参基因必须在给定的实验条件下稳定表达。为促进基因表达研究,获得更准确的表达量数据,筛选相对稳定的内参基因是必要的基础工作。理想的内参基因要求不受外界任何环境因素影响,在不同实验条件下均能稳定表达。但是,大量研究表明,不存在绝对稳定表达的基因(xun,z et al.,.selection and evaluation of reference genes forexpression analysis using qrt
‑
pcr in the beet armyworm spo
‑
doptera exigua (lepidoptera:noctuidae)plos one,2014,9(1):e84730. doi:10.1371/journal.pone.0084730.;shakeel,m.,et al.,gene expression studies of referencegenes for quantitative real
‑
time pcr:an overview in insects.biotechnology letters,2017. 40(2):p.227
‑
236.)。目前,常用的昆虫内参基因有:18s核糖体rna(18s ribosomal rna, 18s rrna)、延伸因子1(elongationfactor
‑
1alpha,ef1
‑
a)、β
‑
肌动蛋白基因(beta actin,β
‑
actin)、28s核糖体rna(28s ribosomal rna,28s rrna)、β
‑
微管蛋白(beta tubulin)、琥珀酸脱氢酶复合体a亚基(succinate dehydrogenase complex subunita,sdha)核糖体蛋白s27 ribosomal protein s27(rsp27)、甘油醛
‑3‑
磷酸脱氢酶(gapdh)和tata盒结合蛋白(tata
‑
box binding protein)等(xun,z et al.,.selection and evaluation of referencegenes for expression analysis using qrt
‑
pcr in the beet armyworm spo
‑
doptera exigua (lepidoptera:noctuidae)plos one,2014,9(1):e84730. doi:10.1371/journal.pone.0084730.;shakeel,m.,et al.,gene expression studies of referencegenes for quantitative real
‑
time pcr:an overview in insects.biotechnology letters,2017. 40(2):p.227
‑
236.)这些基因在细胞中参与正常的生命代谢。在昆虫样本体内,同一基因在不同种类昆虫中的功能、表达水平存在差异。同样,候选内参基因的表达情况也会因为不同昆虫物种或不同实验条件而存在差异。随着qrt
‑
pcr技术的广泛应用,研究发现,昆虫样品种类和实验条件的不同,持家基因的表达量在某些特定条件下会发生较大的变化,如果盲目地借鉴,可能影响最终定量结果的准确性。sa
‑
hel(解旋酶,sa
‑
helicase) 一种广泛存在于多种生物体内的,可以解开氢键的酶,一般在dna或rna复制过程中起到催化双链dna或rna解旋的作用。解旋酶属于分子伴侣,对于确保dna或rna 的正确折叠以及保存和修饰其特定的二级、三级结构必不可少。sa
‑
hel基因是在生物体内非常保守持家基因。通过定量pcr检测表明该基因在麦长管蚜的生长发育的各个时期以及有翅蚜和无翅蚜体内均能稳定表达,为研究麦长管蚜功能基因及在不同发育时期、有翅蚜和无翅蚜体内的表达研究提供了非常理想的内参基因。rpl14(核糖体蛋白质, ribosomal protein l14)是组成核糖体的主要成分,在细胞内蛋白质生物合成中发挥重要作用。核糖体蛋白质广泛分布于各种组织中,它们与核糖核酸共同组成核糖体,在蛋白质的生物合成中起重要作用。越来越多的证据表明:许多种核糖体蛋白除组成核糖体,参与蛋白质的生物合成之外,还具有独立于蛋白质生物合成作用。rpl14基因是在生物体内非常保守持家基因。通过定量pcr检测表明该基因在麦长管蚜的体内均能稳定表达,为研究麦长管蚜功能基因及不同密度处理下的基因表达研究提供了非常理想的内参基因。 sa
‑
rpl11(解旋酶,sa
‑
rpl11icase)一种广泛存在于多种生物体内的,可以解开氢键的酶,一般在dna或rna复制过程中起到催化双链dna或rna解旋的作用。解旋酶属于分子伴侣,对于确保dna或rna的正确折叠以及保存和修饰其特定的二级、三级结构必不可少。sa
‑
rpl11基因是在生物体内非常保守持家基因。通过定量pcr检测表明该基因在麦长管蚜的体内均能稳定表达,为研究麦长管蚜功能基因及信息素e
‑
β
‑
法尼烯溶液处理下的基因表达研究提供了非常理想的内参基因。核糖体(ribosomal)是细胞合成蛋白质的唯一场所。核糖体rna(ribosomal rna,rrna)是组成核糖体的主要成分,是核糖体的结构性或功能性成分。在高级真核生物通常根据其沉降系数来命名为,包括 28s,18s及5s。28s核糖体rna(28s ribosomal rna)广泛存在于多种真核生物体内。 sa
‑
28s基因是在生物体内非常保守持家基因。通过定量pcr检测表明该基因在麦长管蚜的体内均能稳定表达,为研究麦长管蚜功能基因及杀虫剂或抗生素处理下的基因表达研究提供了非常理想的内参基因。
技术实现要素:
6.本发明提供麦长管蚜不同龄期和不同翅型稳定表达的内参基因sa
‑
hel,不同密度处理下稳定表达的内参基因sa
‑
rpl14,信息素e
‑
β
‑
法尼烯溶液处理下稳定表达的内参基因sa
‑
sa
‑
rpl11,杀虫剂处理或抗生素处理下稳定表达内参基因sa
‑
28s的部分序列、克隆方法及应用。
7.本发明同时提供克隆麦长管蚜sa
‑
hel、sa
‑
rpl14、sa
‑
sa
‑
rpl11、sa
‑
28s的特异性引物,建立了基于sybr green1染料技术的rt
‑
pcr方法,从而为麦长管蚜的 sa
‑
hel、sa
‑
rpl14、sa
‑
rpl11、sa
‑
28s作为内参基因,在利用qpcr对麦长管蚜功能基因或作为不同发育时期基因表达,麦蚜翅型分化研究,或作为不同密度处理,或作为信息素e
‑
β
‑
法尼烯溶液处理,或作为杀虫剂或抗生素处理下研究中提供有用的方法。
8.本发明提供的技术方案是:麦长管蚜基因片段,所述基因片为sa
‑
hel基因片段、 sa
‑
rpl14基因片段、sa
‑
rpl11基因片段或sa
‑
28s基因片段,其核苷酸序列分别如seqid no.1、seq id no.6、seq id no.11、seq id no.16所示。在对以上基因序列进行充分分析研究的基础上,避免特定温度下的颈环结构和非特异扩增,并保证符合条件的扩增效率,经过设计筛选得到以上基因片段。
9.本发明还提供一种麦长管蚜基因部分序列的克隆方法:提取麦长管蚜基因组总rna,以试剂盒自带混合引物为反应引物进行rt反应,以产物第一链cdna作为模板、分别以引物对sa
‑
hel
‑
f/sa
‑
hel
‑
r、sa
‑
rpl14
‑
f/sa
‑
rpl14
‑
r、sa
‑
rpl11
‑
f/sa
‑
rpl11
‑
r、 sa
‑
28s
‑
f/sa
‑
28s
‑
r进行rt
‑
pcr扩增,得到阳性克隆,最后经测序验证,其中所述引物对序列如下:
10.sa
‑
hel
‑
f:5
’‑
actggtttgaacgaatatg
‑3’
;
11.sa
‑
hel
‑
r:5
’‑
catccaaatagtgtgtaaga
‑3’
;
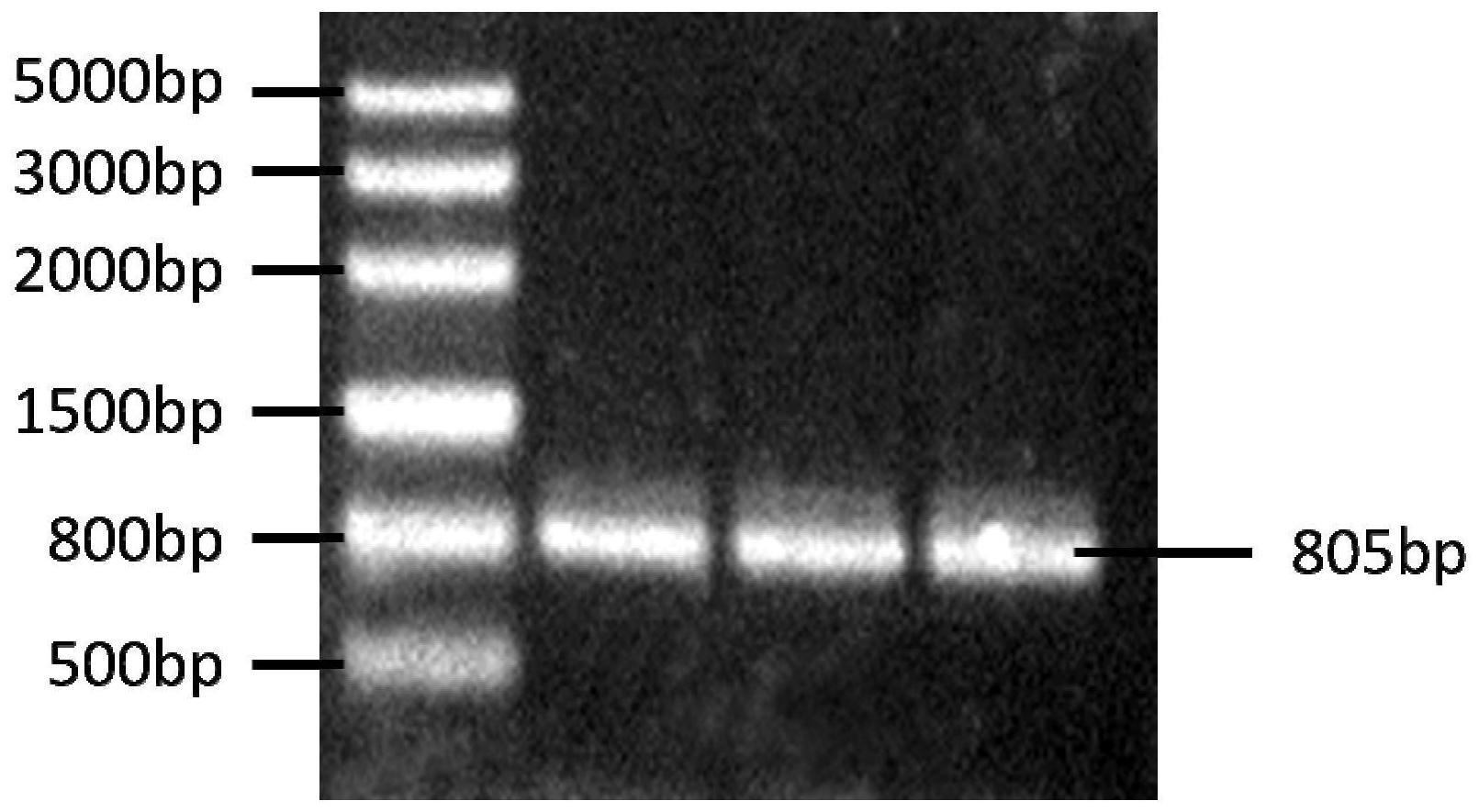
12.所获得目的片段大小为805bp。
13.sa
‑
rpl14
‑
f:5
’‑
aatgtgaggttatgattgtc
‑3’
;
14.sa
‑
rpl14
‑
r:5
’‑
ttaaacagtgcttggtag
‑3’
;
15.所获得目的片段大小为500bp。
16.sa
‑
rpl11
‑
f:5
’‑
tcgtgttgtttcgcttcgtc
‑3’
;
17.sa
‑
rpl11
‑
r:5
’‑
tgatacccaaatcgatgtgttcttg
‑3’
;
18.所获得目的片段大小为418bp。
19.sa
‑
28s
‑
f:5
’‑
cgagtgagccagaaacacat
‑3’
;
20.sa
‑
28s
‑
r:5
’‑
atccccagtctttggcctttt
‑3’
;
21.所获得目的片段大小为617bp。
22.上述的克隆方法,所述pcr扩增的反应体系为:10
×
pcr buffer 2.5μl,2.5mmol/l dntp 2.0μl,25.0mmol/l mgcl
2 2.0μl,5.0u/μl taqdna聚合酶0.5μl,10.0μm上、下游引物各1.0μl,cdna 2.5μl,ddh2o补足至25μl。反应条件如下:94℃预变性5min 后;94℃变性30sec,60℃退火30sec,每个循环降0.5℃,72℃延伸1mim,共30个循环; 94℃再变性30sec,45℃再退火30sec,72℃再延伸1mim,共10个循环;最后于72℃补平7min,终止温度为8℃。
23.本发明还提供一种qpcr扩增麦长管蚜基因部分序列的方法:提取麦长管蚜基因组总rna,以试剂盒自带混合引物为反应引物进行rt反应,以产物第一链cdna作为模板进行荧光定量pcr扩增,荧光染料为sybr green 1,其中定量pcr扩增中的麦长管蚜的定量引物,其
序列为:
24.qsa
‑
hel
‑
f:5
’‑
tgctaccggatgtgggaaaa
‑3’
;
25.qsa
‑
hel
‑
r:5
’‑
tccagccacgttctctgttt
‑3’
;
26.所获得的目的片段大小为152bp。
27.qsa
‑
rpl14
‑
f:5
’‑
aagcaccacagttccaagcg
‑3’
;
28.qsa
‑
rpl14
‑
r:5
’‑
actcggccagtttccacaaa
‑3’
;
29.所获得的目的片段大小为116bp。
30.qsa
‑
rpl11
‑
f:5
’‑
cgtgttgtttcgcttcgtct
‑3’
;
31.qsa
‑
rpl11
‑
r:5
’‑
ctttaagtcggcgaggacca
‑3’
;
32.所获得的目的片段大小为91bp。
33.qsa
‑
28s
‑
f:5
’‑
tctatcaaccggcaaccaca
‑3’
;
34.qsa
‑
28s
‑
r:5
’‑
tggcgaatctcgttgcattt
‑3’
;
35.所获得的目的片段大小为109bp。
36.所述荧光定量pcr扩增的反应条件如下:95℃预变性15min后;先运行40个循环的95℃变性10sec,60℃退火32sec,再运行溶解曲线阶段的95℃15sec,60℃1min,95℃ 30sec,60℃15sec。
37.本发明麦长管蚜sa
‑
hel基因片段可作为内参基因,应用于麦长管蚜功能基因或不同发育时期基因表达,以及有翅蚜和无翅蚜体内基因表达定量pcr检测的应用。
38.麦长管蚜sa
‑
rpl14基因片段可作为内参基因,应用于麦长管蚜功能基因或不同密度处理下基因表达定量pcr检测的应用。
39.麦长管蚜sa
‑
rpl11基因片段可作为内参基因,应用于麦长管蚜功能基因或信息素 e
‑
β
‑
法尼烯溶液处理下基因表达的定量pcr检测。
40.麦长管蚜sa
‑
28s基因片段可作为内参基因,应用于麦长管蚜功能基因或杀虫剂或抗生素处理下基因表达定量pcr检测的应用。
41.本发明根据麦长管蚜转录组测序结果,分析注释得到sa
‑
hel基因、sa
‑
rpl14基因、sa
‑
rpl11基因或sa
‑
28s基因的核苷酸序列,克隆了麦长管蚜的sa
‑
hel、sa
‑
rpl14 基因、sa
‑
rpl11基因或sa
‑
28s基因持家基因的部分序列。设计相应的特异性的定量引物,引物设计过程中在对以上基因序列进行充分分析研究的基础上,避免颈环结构和非特异扩增,并保证引物的扩增效率符合条件。建立基于sybr green1染料技术的rt
‑
pcr 方法,从而为麦长管蚜的sa
‑
hel、sa
‑
rpl14、sa
‑
rpl11或sa
‑
28s作为内参基因,在利用qpcr对麦长管蚜功能基因或作为不同发育时期基因表达,或麦蚜翅型分化研究,或作为不同密度处理,或作为信息素e
‑
β
‑
法尼烯溶液处理,或作为杀虫剂或抗生素处理下研究中提供有用的方法。
42.与现有技术相比,本发明具有以下优点:
43.1.本发明首次克隆得到麦长管蚜的sa
‑
hel基因片段、sa
‑
rpl14基因、sa
‑
rpl11 基因或sa
‑
28s基因;
44.2.本发明首次提出在麦长管蚜定量pcr检测中建立通用的内参基因;
45.3.本发明首次提出将麦长管蚜的sa
‑
hel基因作为内参基因在麦长管蚜不同发育时期的定量pcr检测;首次提出将麦长管蚜的sa
‑
rpl14基因作为内参基因在麦长管蚜不同密度处理的定量pcr检测;首次提出将麦长管蚜的sa
‑
rpl11基因作为内参基因在麦长管蚜
信息素e
‑
β
‑
法尼烯溶液处理下的定量pcr检测;首次提出将麦长管蚜的sa
‑
28s基因作为内参基因在麦长管蚜杀虫剂处理下的定量pcr检测;
46.4.本发明首次提出将麦长管蚜的sa
‑
hel基因作为内参基因可以相对定量有翅蚜和无翅蚜中的基因相对表达水平;
47.5.本发明首次提出将麦长管蚜的sa
‑
28s基因作为内参基因在麦长管蚜抗生素处理下的定量pcr检测;
48.6.本发明提出的检测引物具有特异性,优化了pcr扩增程序,大大的提高了检测效率、缩短了检测时间,提高了检测结果的可信度。
附图说明
49.图1为麦长管蚜sa
‑
hel内参基因的pcr电泳图;m为dna分子量标准;
50.图2为麦长管蚜sa
‑
hel内参基因的熔解曲线;
51.图3为麦长管蚜sa
‑
hel内参基因的标准曲线;
52.图4为sa
‑
hel内参基因在麦长管蚜不同时期的相对表达量。
53.图5为麦长管蚜sa
‑
rpl14内参基因的pcr电泳图;m为dna分子量标准;
54.图6为麦长管蚜sa
‑
rpl14内参基因的熔解曲线;
55.图7为麦长管蚜sa
‑
rpl14内参基因的标准曲线;
56.图8为sa
‑
rpl14内参基因在麦长管蚜不密度处理的相对表达量。
57.图9为麦长管蚜sa
‑
rpl11内参基因的pcr电泳图;m为dna分子量标准;
58.图10为麦长管蚜sa
‑
rpl11内参基因的熔解曲线;
59.图11为麦长管蚜sa
‑
rpl11内参基因的标准曲线。
60.图12为麦长管蚜sa
‑
28s内参基因的pcr电泳图;m为dna分子量标准;
61.图13为麦长管蚜sa
‑
28s内参基因的熔解曲线;
62.图14为麦长管蚜sa
‑
28s内参基因的标准曲线;
63.图15为sa
‑
28s内参基因在麦长管蚜杀虫剂处理下的相对表达量。
具体实施方式
64.下面通过具体实施方式的详细描述来进一步阐明本发明,但并不是对本发明的限制,仅仅作示例说明。下列实施例中未注明具体条件的实验方法,通常按照常规条件。
65.实施例1麦长管蚜sa
‑
hel基因、sa
‑
rpl14基因、sa
‑
rpl11基因或sa
‑
28s基因的克隆
66.1.麦长管蚜总rna提取:
67.利用trizol(amion,usa)提取麦长管蚜的总rna,具体方法如下:取30mg麦长管蚜无翅成虫样品,在液氮中充分研磨,然后快速加入1mltrizol裂解液,充分振荡20s,静止5
‑
10分钟;加入200μl氯仿,振荡20s,静止5分钟,在4℃下12,000rpm离心15min;取上清,加到含有2.5倍体积无水乙醇的离心管内,颠倒混匀,静止10min,
‑
80℃冰箱保存2
‑
4小时;取出样品在4℃下7500rpm离心10min;弃上清,加入1ml 75%的乙醇,清洗沉淀两次,在4℃下12,000rpm离心10min;加入30μl rnase
‑
free water溶解沉淀;利用1%琼脂糖凝胶电泳检测rna的完整性,用超微量紫外分光光度计beckman du 700检测所提总 rna的浓度和质量,质量参数1.9﹤a260/280﹤2.1并且2.0﹤a260/230﹤2.4的rna样品为合格样品,置
‑
80℃冰箱保存
备用。
68.2.反转录
69.利用takara primescript 1st strand cdna合成试剂盒进行反转录,具体操作步骤如下:取1μg麦长管蚜总rna加入无菌pcr管中,加入1μloligo dt primer、1μldntpmixture,用去除rnase的灭菌去离子水补至10μl;混合均匀后置65℃,加热5min后,置冰上迅速冷却;向10μl反应液中加入4μl 5
×
primescript buffer、0.5μl rnase inhibitor、 1.0μl primescript rtase、4.5μl rnase free h2o,在pcr仪上进行反转录反应,反应程序为42℃温浴60min后95℃失活5min,反应结束后放置冰上。合成的第一链cdna置于
‑
20℃短期保存或者
‑
80℃长期保存。
70.3.pcr扩增引物序列的设计
71.sa
‑
hel的上游引物(seq id no.2):5
’‑
actggtttgaacgaatatg
‑3’
;
72.sa
‑
hel的下游引物(seq id no.3):5
’‑
catccaaatagtgtgtaaga
‑3’
。
73.sa
‑
rpl14的上游引物(seq id no.7):5
’‑
aatgtgaggttatgattgtc
‑3’
;
74.sa
‑
rpl14的下游引物(seq id no.8):5
’‑
ttaaacagtgcttggtag
‑3’
;
75.sa
‑
rpl11的上游引物(seq id no.12):5
’‑
tcgtgttgtttcgcttcgtc
‑3’
;
76.sa
‑
rpl11的下游引物(seq id no.13):5
’‑
tgatacccaaatcgatgtgttcttg
‑3’
;
77.sa
‑
28s的上游引物(seq id no.17):5
’‑
cgagtgagccagaaacacat
‑3’
;
78.sa
‑
28s的下游引物(seq id no.8):5
’‑
atccccagtctttggcctttt
‑3’
。
79.从麦长管蚜转录组中获得麦长管蚜hel、sa
‑
rpl14、sa
‑
rpl11、sa
‑
28s候选内参基因的核酸部分序列。上述引物利用unafold在线软件(http://mfold.rna.albany.edu/?q =mfold/dna
‑
folding
‑
form)分析基因核酸部分序列的二级结构,软件设置如下:meltingtemperature,60℃;dna sequence,linear;na+concentration,50mm;mg2+concentration, 3mm;其它选项为初始设置。获得基因模板序列含有茎环结构的位点后,利用 ncbi
‑
primer
‑
blast在线软件(http://www.ncbi.nlm.nih. gov/tools/primer
‑
blast/index.cgi?link_loc=blasthome)设计引物,软件设置如下:primermelting temperature,57
‑
63℃;primer gc content,40
‑
60%;pcr product size,500
‑
1500 base pairs;excluded regions,茎环结构位点;其它选项为初始设置。
80.4.聚合酶链式反应(pcr)
81.以rt得到的第一链cdna为模板进行pcr扩增。反应体系为:10
×
pcr buffer 2.5μl, 2.5mmol/l dntp 2.0μl,25.0mmol/l mgcl
2 2.0μl,5.0u/μl taqdna聚合酶0.5μl, 10.0μm上、下游引物各1.0μl,cdna 2.5μl,ddh2o补足至25μl。反应条件如下:94℃预变性5min后;94℃变性30sec,60℃退火30sec,每个循环降0.5℃,72℃延伸1mim,共30个循环;94℃再变性30sec,45℃再退火30sec,72℃再延伸1mim,共10个循环;最后于72℃补平7min,终止温度为8℃。pcr产物经1%琼脂糖凝胶电泳检测目的片段 (如图1、图5、图9、图12所示)。
82.5.目的基因片段获得与序列测定
83.经琼脂糖凝胶电泳后,对以上pcr产物进行胶回收,经回收纯化的dna片段分别链接到载体pmd 17
‑
t vevtor上,转化到大肠杆菌dh5α感受态细胞中,经amp抗生素筛选获得确认已插入目的片段的阳性克隆菌株,进行核苷酸序列测定,分别得到麦长管蚜sa
‑
hel、sa
‑
rpl14、sa
‑
rpl11、sa
‑
28s的核苷酸序列,具体序列分别见seq id no.1、 seq id no.6、
seqidno.11、seqidno.16。
84.实施例2荧光定量pcr扩增引物的设计与合成
85.根据麦长管蚜ncbi及实施例1中克隆的各基因片段序列,设计基因的荧光定量pcr的引物。利用unafold在线软件(http://mfold.rna.albany.edu/?q=mfold/dna
‑
folding
‑
form)分析各基因核酸部分序列的二级结构,软件设置如下:meltingtemperature,60℃;dnasequence,linear;na+concentration,50mm;mg
2+
concentration,3mm;其它选项为初始设置。获得各基因模板序列含有茎环结构的位点后,利用ncbi
‑
primer
‑
blast在线软件(http://www.ncbi.nlm.nih.gov/tools/primer
‑
blast/index.cgi?link_loc=blasthome)设计引物,软件设置如下:primermeltingtemperature,57
‑
63℃;primergccontent,40
‑
60%;pcrproductsize,150
‑
300basepairs;excludedregions,茎环结构位点;其它选项为初始设置。针对每个基因序列设计多对引物,首先利用普通pcr扩增,筛选可以得到单一条带的应该定量引物,在进行荧光定量pcr实验。各基因的荧光定量pcr的引物如下:
86.qsa
‑
hel
‑
f(seqidno.4):5
’‑
tgctaccggatgtgggaaaa
‑3’
;
87.qsa
‑
hel
‑
r(seqidno.5):5
’‑
tccagccacgttctctgttt
‑3’
。
88.qsa
‑
rpl14
‑
f(seqidno.9):5
’‑
aagcaccacagttccaagcg
‑3’
;
89.qsa
‑
rpl14
‑
r(seqidno.10):5
’‑
actcggccagtttccacaaa
‑3’
;
90.qsa
‑
rpl11
‑
f(seqidno.14):5
’‑
cgtgttgtttcgcttcgtct
‑3’
;
91.qsa
‑
rpl11
‑
r(seqidno.15):5
’‑
ctttaagtcggcgaggacca
‑3’
;
92.qsa
‑
28s
‑
f(seqidno.19):5
’‑
tctatcaaccggcaaccaca
‑3’
;
93.qsa
‑
28s
‑
r(seqidno.20):5
’‑
tggcgaatctcgttgcattt
‑3’
。
94.实施例3sa
‑
hel基因在麦长管蚜有翅成蚜和无翅成蚜中的荧光定量pcr检测
95.1.麦长管蚜有翅成蚜和无翅成蚜的总rna提取:
96.利用trizol(amion,usa)分别提取麦长管蚜有翅成蚜和无翅成蚜的总rna,具体方法同实施例1中提取麦长管蚜的总rna方法。
97.2.反转录
98.利用takaraprimescript1ststrandcdna合成试剂盒进行反转录,具体操作步骤如下:取1μg麦长管蚜总rna加入无菌pcr管中,加入1μloligodtprimer、1μldntpmixture,用去除rnase的灭菌去离子水补至10μl;混合均匀后置65℃,加热5min后,置冰上迅速冷却;向10μl反应液中加入4μl5
×
primescriptbuffer、0.5μlrnaseinhibitor、1.0μlprimescriptrtase、4.5μlrnasefreeh2o,在pcr仪上进行反转录反应,反应程序为42℃温浴60min后95℃失活5min,反应结束后放置冰上。合成的第一链cdna置于
‑
20℃短期保存或者
‑
80℃长期保存。
99.3.荧光定量pcr扩增引物合成
100.荧光定量pcr上游引物(seqidno.4):5
’‑
tgctaccggatgtgggaaaa
‑3’
;
101.荧光定量pcr下游引物(seqidno.5):5
’‑
tccagccacgttctctgttt
‑3’
。
102.4.荧光定量pcr
103.以rt得到的第一链cdna为模板进行荧光定量pcr检测。基于sybrgreeni染料的qpcr的反应体系为:ssofastevagreensupermix10μl、的上、下游引物(10.0μm)各1.0μl,
cdna 1.0μl,ddh2o补足至20μl。所述荧光定量pcr扩增的反应条件如下:95℃预变性15min后;先运行40个循环的95℃变性10sec,60℃退火32sec,再运行溶解曲线阶段的 95℃15sec,60℃1min,95℃30sec,60℃15sec。
104.5.引物的扩增效率
105.使用麦长管蚜无翅成虫cdna进行一系列梯度稀释后作为模板,进行实时荧光定量 pcr,制作引物的扩增效率标准曲线。引物扩增效率计算方式如下:扩增效率(e)=(10
[
‑
1/斜率]
‑
1)
×
100。
[0106]
6.实验结果
[0107]
本发明按照定量pcr反应程序将麦长管蚜的有翅成蚜和无翅成蚜的基因组rna样品进行定量pcr扩增,获得熔解曲线(melting curve)(图2)、定量循环(quantificationcycle,cq)和扩增曲线(amplification plot)。结合熔解曲线判断扩增产物的特异性,理想的熔解曲线应该为单峰曲线。从本实验的引物熔解曲线分析得出,麦长管蚜有翅成蚜和无翅成蚜样品的sa
‑
hel基因检测显现单峰曲线,无引物二聚体等非特异性扩增产生,说明本发明涉及的荧光定量pcr扩增引物(seq id no.4和5)扩增条带单一,特异性强,没有非特异性扩增出现。本发明从麦长管蚜有翅有翅成蚜和无翅成蚜的荧光定量pcr 扩增曲线可以看出,该对引物在模板的不同稀释浓度下进行扩增,产物扩增量都经历了典型的基线、指数增长期、线性期和平台期等扩增过程,说明该基因可以用于扩增麦长管蚜有翅有翅成蚜和无翅成蚜体内的sa
‑
hel基因。将麦长管蚜成虫cdna进行一系列 5倍梯度稀释后作为模板,进行实时荧光定量pcr检测,获得一系列模板的cq值,以 cq值为纵坐标,以稀释倍数为横坐标绘制麦长管蚜sa
‑
hel基因的拟合曲线(如图3) 为:y=
‑
3.2493x+31.709,相关系数(r2)为0.994,将拟合曲线的斜率带入引物扩增效率计算方式:扩增效率(e)=(10
[
‑
1/斜率]
‑
1)
×
100,计算获得其扩增效率为103%,该引物的扩增效率在100%左右,说明该引物的扩增效率很高,符合作为麦长管蚜有翅成蚜和无翅成蚜的荧光定量pcr检测的内参基因。
[0108]
实施例4sa
‑
hel基因在麦长管蚜不同发育时期的荧光定量pcr检测
[0109]
1.麦长管蚜总rna提取:
[0110]
分别提取麦长管蚜1龄若虫、2龄若虫、3龄若虫、4龄若虫和成虫等不同发育时期的总rna,收集不同发育时期麦长管蚜样品:每管收集约40头1龄若虫,30头2龄若虫,20 头3龄若虫,20头4龄若虫,20头无翅成蚜,每一发育时期均收集4管,编号后放入液氮中速冻,于
‑
80℃冰箱短期保存。rna方法如下:取30mg样品,在液氮中充分研磨,然后快速加入1mltrizol裂解液,充分振荡20s,静止5
‑
10分钟;加入200μl氯仿,振荡20s,静止5分钟,在4℃下12,000rpm离心15min;取上清,加到含有2.5倍体积无水乙醇的离心管内,颠倒混匀,静止10min,
‑
80℃冰箱保存2
‑
4小时;取出样品在4℃下7500rpm离心10min;弃上清,加入1ml 75%的乙醇,清洗沉淀两次,在4℃下12,000rpm离心10min;加入30μl rnase
‑
free water溶解沉淀;利用1%琼脂糖凝胶电泳检测rna的完整性,用超微量紫外分光光度计beckman du 700检测所提总rna的浓度和质量,质量参数1.9﹤a260/280﹤2.1 并且2.0﹤a260/230﹤2.4的rna样品为合格样品,置
‑
80℃冰箱保存备用。
[0111]
2.反转录
[0112]
利用takara primescript 1st strand cdna合成试剂盒进行反转录,具体操作步骤同实施例3中的反转录。
[0113]
3.荧光定量pcr扩增引物合成
[0114]
荧光定量pcr上游引物(seq id no.4):5
’‑
tgctaccggatgtgggaaaa
‑3’
;
[0115]
荧光定量pcr下游引物(seq id no.5):5
’‑
tccagccacgttctctgttt
‑3’
。
[0116]
4.荧光定量pcr
[0117]
以rt得到的第一链cdna为模板进行荧光定量pcr检测。基于sybr green i染料的 qpcr的反应体系为:ssofast evagreen supermix 10μl、的上、下游引物(10.0μm)各1.0μl, cdna 1.0μl,ddh2o补足至20μl。所述荧光定量pcr扩增的反应条件如下:95℃预变性 15min后;先运行40个循环的95℃变性10sec,60℃退火32sec,再运行溶解曲线阶段的 95℃15sec,60℃1min,95℃30sec,60℃15sec。
[0118]
5.实验结果
[0119]
本发明按照定量pcr反应程序将麦长管蚜的1龄若虫、2龄若虫、3龄若虫、4龄若虫和成虫等不同发育时期的基因组rna样品进行定量pcr扩增,获得熔解曲线(meltingcurve)、定量循环(quantification cycle,cq)和扩增曲线(amplification plot)。结合熔解曲线判断扩增产物的特异性,理想的熔解曲线应该为单峰曲线。从本实验的引物熔解曲线分析得出,麦长管蚜1龄若虫、2龄若虫、3龄若虫、4龄若虫和成虫等不同发育时期样品的sa
‑
hel基因检测显现单峰曲线,无引物二聚体等非特异性扩增产生,说明本发明涉及的荧光定量pcr扩增引物(seq id no.4和5)扩增条带单一,特异性强,没有非特异性扩增出现。分析sa
‑
hel基因在麦长管蚜不同发育时期样品中表达情况(图 4),统计分析sa
‑
hel基因在麦长管蚜1龄若虫、2龄若虫、3龄若虫、4龄若虫和成虫各样品中的cq值,发现sa
‑
hel基因在麦长管蚜不同发育时期能稳定表达,差异不显著(p>0.05),说明sa
‑
hel基因可以用于麦长管蚜1龄若虫、2龄若虫、3龄若虫、4 龄若虫和成虫等不同发育时期样品体内的功能基因荧光定量pcr检测的内参基因。
[0120]
实施例5sa
‑
rpl14基因在麦长管蚜不同密度处理下的荧光定量pcr检测
[0121]
1.麦长管蚜的总rna提取:
[0122]
将麦长管蚜的3龄若蚜按照1头/皿,30头/皿,60头/皿进行不同密度处理,处理24h后收集蚜虫样品,利用trizol(amion,usa)分别提取各处理下麦长管蚜的总rna,具体方法同实施例1。
[0123]
2.反转录
[0124]
利用takara primescript 1st strand cdna合成试剂盒进行反转录,具体操作步骤同实施例3中的反转录。
[0125]
3.荧光定量pcr扩增引物合成
[0126]
荧光定量pcr上游引物(seq id no.9):5
’‑
aagcaccacagttccaagcg
‑3’
;
[0127]
荧光定量pcr下游引物(seq id no.10):5
’‑
actcggccagtttccacaaa
‑3’
。
[0128]
4.荧光定量pcr
[0129]
以rt得到的第一链cdna为模板进行荧光定量pcr检测。基于sybr green i染料的 qpcr的反应体系为:ssofast evagreen supermix 10μl、的上、下游引物(10.0μm)各1.0μl, cdna 1.0μl,ddh2o补足至20μl。所述荧光定量pcr扩增的反应条件如下:95℃预变性 15min后;先运行40个循环的95℃变性10sec,60℃退火32sec,再运行溶解曲线阶段的 95℃15sec,60℃1min,95℃30sec,60℃15sec。
cdna 1.0μl,ddh2o补足至20μl。所述荧光定量pcr扩增的反应条件如下:95℃预变性 15min后;先运行40个循环的95℃变性10sec,60℃退火32sec,再运行溶解曲线阶段的 95℃15sec,60℃1min,95℃30sec,60℃15sec。
[0144]
5.引物的扩增效率
[0145]
使用麦长管蚜无翅成虫cdna进行一系列梯度稀释后作为模板,进行实时荧光定量 pcr,制作引物的扩增效率标准曲线。引物扩增效率计算方式如下:扩增效率(e)=(10
[
‑
1/斜率]
‑
1)
×
100。
[0146]
6.实验结果
[0147]
本发明按照定量pcr反应程序将麦长管蚜的有翅成蚜和无翅成蚜的基因组rna样品进行定量pcr扩增,获得熔解曲线(melting curve)(如图10所示)、定量循环 (quantification cycle,cq)和扩增曲线(amplification plot)。结合熔解曲线判断扩增产物的特异性,理想的熔解曲线应该为单峰曲线。从本实验的引物熔解曲线分析得出,麦长管蚜有翅成蚜和无翅成蚜样品的sa
‑
rpl11基因检测显现单峰曲线,无引物二聚体等非特异性扩增产生,说明本发明涉及的荧光定量pcr扩增引物(seq id no.14和15) 扩增条带单一,特异性强,没有非特异性扩增出现。本发明从麦长管蚜有翅有翅成蚜和无翅成蚜的荧光定量pcr扩增曲线可以看出,该对引物在模板的不同稀释浓度下进行扩增,产物扩增量都经历了典型的基线、指数增长期、线性期和平台期等扩增过程,说明该基因可以用于扩增麦长管蚜有翅有翅成蚜和无翅成蚜体内的sa
‑
rpl11基因。将麦长管蚜成虫cdna进行一系列5倍梯度稀释后作为模板,进行实时荧光定量pcr检测,获得一系列模板的cq值,以cq值为纵坐标,以稀释倍数为横坐标绘制麦长管蚜sa
‑
rpl11 基因的拟合曲线(如图11所示)为:y=
‑
3.558x+37.779,相关系数(r2)为0.9997,将拟合曲线的斜率带入引物扩增效率计算方式:扩增效率(e)=(10
[
‑
1/斜率]
‑
1)
×
100,计算获得其扩增效率为91%,该引物的扩增效率在100%左右,说明该引物的扩增效率很高,符合作为麦长管蚜荧光定量pcr检测的内参基因。分析sa
‑
sa
‑
rpl11基因在麦长管蚜信息素e
‑
β
‑
法尼烯溶液处理样品中表达情况,统计分析sa
‑
sa
‑
rpl11基因在麦长管蚜信息素e
‑
β
‑
法尼烯溶液处理样品和对照组中的cq值,发现sa
‑
sa
‑
rpl11基因在麦长管蚜信息素e
‑
β
‑
法尼烯溶液处理样品中能稳定表达,差异不显著(p>0.05),说明sa
‑
sa
‑
rpl11 基因可以用于麦长管信息素e
‑
β
‑
法尼烯溶液处理样品体内的功能基因荧光定量pcr检测的内参基因。
[0148]
实施例7sa
‑
28s基因在麦长管蚜抗生素处理下的荧光定量pcr检测
[0149]
1.麦长管蚜的总rna提取:
[0150]
将麦长管蚜的成虫用50μg/ml利福平处理24h后收集蚜虫样品,利用trizol(amion, usa)分别提取各处理下麦长管蚜的总rna,具体方法同实施例1。
[0151]
2.反转录
[0152]
利用takara primescript 1st strand cdna合成试剂盒进行反转录,具体操作步骤同实施例3中的反转录。
[0153]
3.荧光定量pcr扩增引物合成
[0154]
荧光定量pcr上游引物(seq id no.19):5
’‑
tctatcaaccggcaaccaca
‑3’
;
[0155]
荧光定量pcr下游引物(seq id no.20):5
’‑
tggcgaatctcgttgcattt
‑3’
。
[0156]
4.荧光定量pcr
[0157]
以rt得到的第一链cdna为模板进行荧光定量pcr检测。基于sybr green i染料的 qpcr的反应体系为:ssofast evagreen supermix 10μl、的上、下游引物(10.0μm)各1.0μl, cdna 1.0μl,ddh2o补足至20μl。所述荧光定量pcr扩增的反应条件如下:95℃预变性 15min后;先运行40个循环的95℃变性10sec,60℃退火32sec,再运行溶解曲线阶段的 95℃15sec,60℃1min,95℃30sec,60℃15sec。
[0158]
5.引物的扩增效率
[0159]
使用麦长管蚜无翅成虫cdna进行一系列梯度稀释后作为模板,进行实时荧光定量 pcr,制作引物的扩增效率标准曲线。引物扩增效率计算方式如下:扩增效率(e)=(10
[
‑
1/斜率]
‑
1)
×
100。
[0160]
6.实验结果
[0161]
本发明按照定量pcr反应程序将麦长管蚜的抗生素处理和对照的基因组rna样品进行定量pcr扩增,获得熔解曲线(melting curve)(图13)、定量循环(quantificationcycle,cq)和扩增曲线(amplification plot)。结合熔解曲线判断扩增产物的特异性,理想的熔解曲线应该为单峰曲线。从本实验的引物熔解曲线分析得出,麦长管蚜抗生素处理的成蚜样品的sa
‑
28s基因检测显现单峰曲线,无引物二聚体等非特异性扩增产生,说明本发明涉及的荧光定量pcr扩增引物(seq id no.19和20)扩增条带单一,特异性强,没有非特异性扩增出现。本发明从麦长管蚜抗生素处理的成蚜的荧光定量pcr扩增曲线可以看出,该对引物在模板的不同稀释浓度下进行扩增,产物扩增量都经历了典型的基线、指数增长期、线性期和平台期等扩增过程,说明该基因可以用于扩增麦长管蚜抗生素处理的成蚜体内的sa
‑
28s基因。将麦长管蚜成虫cdna进行一系列5倍梯度稀释后作为模板,进行实时荧光定量pcr检测,获得一系列模板的cq值,以cq值为纵坐标,以稀释倍数为横坐标绘制麦长管蚜sa
‑
28s基因的拟合曲线(如图14所示)为: y=
‑
3.0672x+30.912,相关系数(r2)为0.9886,将拟合曲线的斜率带入引物扩增效率计算方式:扩增效率(e)=(10
[
‑
1/斜率]
‑
1)
×
100,计算获得其扩增效率为112%,该引物的扩增效率在100%左右,说明该引物的扩增效率很高,符合作为麦长管蚜抗生素处理的成蚜的荧光定量pcr检测的内参基因。
[0162]
实施例8sa
‑
28s基因在麦长管蚜杀虫剂处理下的荧光定量pcr检测
[0163]
1.麦长管蚜的总rna提取:
[0164]
将麦长管蚜的成虫用不同浓度杀虫剂(lc
20
致死率为20%和lc
50
致死率为50%的杀虫剂浓度)处理24h后收集蚜虫样品。所选杀虫剂包括吡虫啉、噻虫嗪和毒死蜱,对麦长管蚜成虫进行生物活性测定,将带麦蚜的麦苗全部浸泡在供试药剂中,停留10s后室温干燥 5
‑
10min,放入9cm培养皿中。以0.1%triton x
‑
100为对照,每个处理包括对照均收集4 管,每管约50头试虫,放入液氮中速冻,于
‑
80℃冰箱短期保存。利用trizol(amion, usa)分别提取各处理下麦长管蚜的总rna,具体方法同实施例1。
[0165]
2.反转录
[0166]
利用takara primescript 1st strand cdna合成试剂盒进行反转录,具体操作步骤同实施例3中的反转录。
[0167]
3.荧光定量pcr扩增引物合成
[0168]
荧光定量pcr上游引物(seq id no4):5
’‑
tctatcaaccggcaaccaca
‑3’
;
[0169]
荧光定量pcr下游引物(seq id no5):5
’‑
tggcgaatctcgttgcattt
‑3’
。
[0170]
4.荧光定量pcr
[0171]
以rt得到的第一链cdna为模板进行荧光定量pcr检测。基于sybr green i染料的 qpcr的反应体系为:ssofast evagreen supermix 10μl、的上、下游引物(10.0μm)各1.0μl, cdna 1.0μl,ddh2o补足至20μl。所述荧光定量pcr扩增的反应条件如下:95℃预变性15min后;先运行40个循环的95℃变性10sec,60℃退火32sec,再运行溶解曲线阶段的 95℃15sec,60℃1min,95℃30sec,60℃15sec。
[0172]
5.引物的扩增效率
[0173]
使用麦长管蚜无翅成虫cdna进行一系列梯度稀释后作为模板,进行实时荧光定量 pcr,制作引物的扩增效率标准曲线。引物扩增效率计算方式如下:扩增效率(e)=(10
[
‑
1/斜率]
‑
1)
×
100。
[0174]
6.实验结果
[0175]
本发明按照定量pcr反应程序将麦长管蚜的有翅成蚜和无翅成蚜的基因组rna样品进行定量pcr扩增,获得熔解曲线(melting curve)、定量循环(quantification cycle, cq)和扩增曲线(amplification plot)。结合熔解曲线判断扩增产物的特异性,理想的熔解曲线应该为单峰曲线。从本实验的引物熔解曲线分析得出,麦长管蚜杀虫剂处理的成蚜样品的sa
‑
28s基因检测显现单峰曲线,无引物二聚体等非特异性扩增产生,说明本发明涉及的荧光定量pcr扩增引物(seq id no4和5)扩增条带单一,特异性强,没有非特异性扩增出现。本发明从麦长管蚜杀虫剂处理的成蚜的荧光定量pcr扩增曲线可以看出,该对引物在模板的不同稀释浓度下进行扩增,产物扩增量都经历了典型的基线、指数增长期、线性期和平台期等扩增过程,说明该基因可以用于扩增麦长管蚜有翅有翅成蚜和无翅成蚜体内的sa
‑
28s基因。将麦长管蚜成虫cdna进行一系列5倍梯度稀释后作为模板,进行实时荧光定量pcr检测,获得一系列模板的cq值,以cq值为纵坐标,以稀释倍数为横坐标绘制麦长管蚜sa
‑
28s基因的拟合曲线为:y=
‑
3.0672x+30.912,相关系数(r2)为0.9886,将拟合曲线的斜率带入引物扩增效率计算方式:扩增效率(e)=(10
[
‑
1/斜率]
‑
1)
×
100,计算获得其扩增效率为112%,该引物的扩增效率在100%左右,说明该引物的扩增效率很高,符合作为麦长管蚜杀虫剂处理的荧光定量pcr检测的内参基因。分析sa
‑
28s基因在麦长管蚜不同杀虫剂浓度处理的样品中表达情况(如图14所示),统计分析sa
‑
28s基因在麦长管在lc
20
、lc
50
和ck中的cq值,发现sa
‑
28s基因在麦长管蚜不同杀虫剂浓度处理下能稳定表达,差异不显著(p>0.05),说明sa
‑
28s基因可以用于麦长管蚜杀虫剂处理样品体内的功能基因荧光定量pcr检测的内参基因。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1