一种反式烯烃的氘代方法
1.本发明涉及有机合成技术领域,具体涉及一种反式烯烃的氘代方法。
背景技术:
2.氘代化合物是一类重要的高附加值化学品,除了作为溶剂应用于核磁共振波谱分析领域外,在探究药物代谢、揭示有机反应机理和改善材料性能等方面也有着重要的应用。氘代反应不仅可用于改进在药代动力学与代谢方面有缺陷的已上市药物,而且已经在药物化学中用于开发新药的结构修饰手段。氘代药物具有巨大的市场价值。2017年,首例氘代药物austedo
tm
(deutetra benazine,氘代丁苯那嗪片)正式被批准上市,它作为囊泡单胺转运蛋白的阻断剂,可以有效治疗亨廷顿舞蹈症引发的异常不主动运动。
3.目前,氘代烯烃的合成主要是通过过渡金属催化炔烃的半氘化合成,氘源一般为氘气或氘水。例如,相关技术中使用d2o作为氘源,在氧化条件下卡宾催化的烯醛的α,γ
‑
氘化。一般用氘气代替氢气进行炔烃的半氘化是可行的,但这种方法最大的缺点是氘气价格昂贵,且官能团耐受性有限。此外,过渡金属催化的烯烃可直接进行氢
‑
氘交换反应。相关技术中还发现ruhcl(co)(pph3)3可以有效地催化与烯烃的h/d交换反应。相关技术中的氘代反应通常对末端烯烃具有较高的氘化选择性,对非末端烯烃的氘代效果较差。
4.因此,需要开发一种高效的反式烯烃的氘代方法。
技术实现要素:
5.为解决现有技术中存在的问题,本发明提供了一种反式烯烃的氘代方法,该方法氘代的效率高。
6.本发明提供了一种反式烯烃的氘代方法,将具有下式(ⅰ)的烯烃化合物和引发剂、氘代试剂以及溶剂反应,制得具有下式(ⅱ)的反式氘代烯烃化合物;
[0007][0008]
其中:r1和r2均独立地选自芳基、取代芳基、杂芳基和取代杂芳基中的一种。
[0009]
根据本发明的一些实施方式,所述芳基为c
20
以下的芳基。
[0010]
根据本发明的一些实施方式,所述芳基为c
10
以下的芳基。
[0011]
根据本发明的一些实施方式,所述芳基包括苯基。
[0012]
根据本发明的一些实施方式,所述取代芳基为烷基芳基、烷氧基芳基、硝基芳基、卤代芳基或羟基芳基。
[0013]
根据本发明的一些实施方式,所述烷基芳基为一取代烷基芳基或二取代烷基芳基。
[0014]
根据本发明的一些实施方式,所述二取代烷基芳基中烷基相同或不同。
[0015]
根据本发明的一些实施方式,所述烷基芳基中烷基为c
1~8
烷基。
[0016]
根据本发明的一些实施方式,所述c
1~8
烷基包括但不限于甲基和叔丁基。
[0017]
根据本发明的一些实施方式,所述烷基芳基包括甲苯基。
[0018]
根据本发明的一些实施方式,所述烷氧基芳基包括甲氧基苯基。
[0019]
根据本发明的一些实施方式,所述卤代芳基为一取代或二取代苯基。
[0020]
根据本发明的一些实施方式,所述卤代为f代、cl代或br代。
[0021]
根据本发明的一些实施方式,所述二取代相同或不同。
[0022]
根据本发明的一些实施方式,所述卤代芳基包括氟苯基、氯苯基或溴苯基。
[0023]
根据本发明的一些实施方式,所述羟基芳基包括苯酚基。
[0024]
根据本发明的一些实施方式,所述杂芳基为c
20
以下的杂芳基。
[0025]
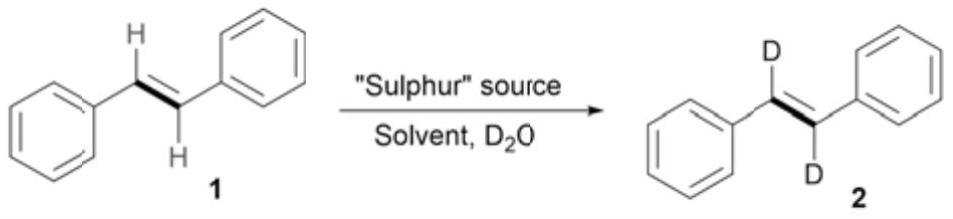
根据本发明的一些实施方式,所述杂芳基为c
10
以下的杂芳基。
[0026]
根据本发明的一些实施方式,所述杂芳基包括噻吩基或吡啶基。
[0027]
根据本发明的一些实施方式,所述取代杂芳基为烷基杂芳基、烷氧基杂芳基或硝基杂芳基。
[0028]
根据本发明的一些实施方式,所述烷基杂芳基为一取代烷基杂芳基或二取代烷基杂芳基。
[0029]
根据本发明的一些实施方式,所述二取代烷基杂芳基中烷基相同或不同。
[0030]
根据本发明的一些实施方式,所述烷基杂芳基中烷基为c
1~8
烷基。
[0031]
根据本发明的一些实施方式,所述c
1~8
烷基包括但不限于甲基和叔丁基。
[0032]
根据本发明的一些实施方式,所述引发剂包括含硫化合物。
[0033]
根据本发明的一些实施方式,所述含硫化合物包括无机硫和有机硫中的至少一种。
[0034]
根据本发明的一些实施方式,所述无机硫包括硫盐和单质硫中的至少一种。
[0035]
根据本发明的一些实施方式,所述硫盐包括硫化钠、硫化钾和硫化铵中的至少一种。
[0036]
根据本发明的一些实施方式,所述有机硫包括黄原酸盐类化合物。
[0037]
根据本发明的一些实施方式,所述黄原酸盐类化合物包括异丙基黄原酸钾、正丁基黄原酸钾、乙基黄原酸钾和乙基黄原酸钠中的至少一种。
[0038]
根据本发明的一些实施方式,所述引发剂为乙基黄原酸钾。
[0039]
根据本发明的一些实施方式,所述氘代试剂为重水。
[0040]
重水作为氘源,生产成本低且无污染。
[0041]
根据本发明的一些实施方式,所述溶剂包括含氮溶剂、含硫溶剂和烃类溶剂中的至少一种。
[0042]
根据本发明的一些实施方式,所述含氮溶剂包括n,n
‑
二甲基甲酰胺、n,n
‑
二甲基乙酰胺和n
‑
甲基吡咯烷酮中的至少一种。
[0043]
根据本发明的一些实施方式,所述烃类溶剂包括甲苯和二甲苯中的至少一种。
[0044]
根据本发明的一些实施方式,所述含硫溶剂包括二甲基亚砜。
[0045]
根据本发明的一些实施方式,所述溶剂为n,n
‑
二甲基甲酰胺。
[0046]
根据本发明的一些实施方式,所述反应的温度为100℃~150℃。
[0047]
根据本发明的一些实施方式,所述反应的温度为150℃。
[0048]
根据本发明的一些实施方式,所述反应的时间为12h~24h。
[0049]
根据本发明的一些实施方式,所述反应的时间为24h。
[0050]
根据本发明的一些实施方式,所述烯烃化合物和所述引发剂的摩尔比为1:0.8~1.2。
[0051]
在该比例下能够得到氘代率较高的烯烃产物,低于该摩尔比所得氘代烯烃的氘代率较低,高于该摩尔比并不会提高氘代烯烃的氘代率,造成原料利用率下降。
[0052]
根据本发明的一些实施方式,所述烯烃化合物和所述氘代试剂的摩尔比为1:50~100。
[0053]
根据本发明至少一个具体的实施方式,具备如下有益效果:
[0054]
本发明以烯烃化合物为原料,无过渡金属催化,在添加引发剂(含硫化合物)的条件下加热搅拌反应;通过一锅法合成了反式氘代烯烃化合物,除终产物外,无需针对转化过程中的中间体(相关技术中使用的过渡金属催化剂、配体及其中间产物)进行分离和纯化,一步完成合成,容易实现产业化生产,为工业生产中减少了资金和劳动力投入。
具体实施方式
[0055]
以下将结合实施例对本发明的构思及产生的技术效果进行清楚、完整地描述,以充分地理解本发明的目的、特征和效果。显然,所描述的实施例只是本发明的一部分实施例,而不是全部实施例,基于本发明的实施例,本领域的技术人员在不付出创造性劳动的前提下所获得的其他实施例,均属于本发明保护的范围。
[0056]
本发明的描述中,参考术语“一个实施例”、“一些实施例”、“示意性实施例”、“示例”、“具体示例”、或“一些示例”等的描述意指结合该实施例或示例描述的具体特征、结构、材料或者特点包含于本发明的至少一个实施例或示例中。在本说明书中,对上述术语的示意性表述不一定指的是相同的实施例或示例。而且,描述的具体特征、结构、材料或者特点可以在任何的一个或多个实施例或示例中以合适的方式结合。
[0057]
下面详细描述本发明的具体实施例。
[0058]
本发明实施方式中硫源起到引发剂的作用。
[0059]
本发明实施方式中分离产率为经过后处理后得到的目标化合物纯品量与理论量之比(转化为百分数)。
[0060]
实施例1
[0061]
本实施例提供了一种反式氘代二苯乙烯的合成方法,合成路线包括如下式所示的步骤:
[0062][0063]
在25ml反应管中,分别加入反式二苯乙烯(90mg,0.5mmo1)、硫源(sulphur source)为硫脲(38mg,0.5mmo1)和重水(0.5ml,0.028mol),以n,n
‑
二甲基甲酰胺(solvent,2ml)为溶剂的条件下,在反应温度为150℃情况下,搅拌反应24h。
[0064]
反应完成后,加入乙酸乙酯进行淬灭反应,再加入饱和食盐水洗涤,分出有机相,水相再用乙酸乙酯反复萃取3次合并有机相,加入无水硫酸钠干燥,经减压蒸馏除去溶剂,再经柱层析分离,分离后得到白色固体(78mg,分离产率85%)。
[0065]
反应完成后,白色固体经核磁共振(nuclear magnetic resonance spectroscopy,nmr)检测,氘代率为0。
[0066]
实施例2
[0067]
本实施例提供了一种反式氘代二苯乙烯的合成方法,与实施例1的差异在于:硫源为硫代乙酰胺(38mg,0.5mmo1)。
[0068]
反应完成后,加入乙酸乙酯进行淬灭反应,再加入饱和食盐水洗涤,分出有机相,水相再用乙酸乙酯反复萃取3次合并有机相,加入无水硫酸钠干燥,经减压蒸馏除去溶剂,再经柱层析分离,分离后得到白色固体(75mg,分离产率82%)。
[0069]
反应完成后,白色固体经核磁共振(nuclear magnetic resonance spectroscopy,nmr)检测,氘代率为0。
[0070]
实施例3
[0071]
本实施例提供了一种反式氘代二苯乙烯的合成方法,与实施例1的差异在于:硫源为异丙基黄原酸钾(87mg,0.5mmo1)。
[0072]
反应完成后,加入乙酸乙酯进行淬灭反应,再加入饱和食盐水洗涤,分出有机相,水相再用乙酸乙酯反复萃取3次合并有机相,加入无水硫酸钠干燥,经减压蒸馏除去溶剂,再经柱层析分离,分离后得到白色固体(75mg,分离产率82%)。
[0073]
反应完成后,白色固体经核磁共振(nuclear magnetic resonance spectroscopy,nmr)检测,氘代率为25%。
[0074]
实施例4
[0075]
本实施例提供了一种反式氘代二苯乙烯的合成方法,与实施例3的差异在于:硫源为正丁基黄原酸钾(79mg,0.5mmo1)。
[0076]
反应完成后,加入乙酸乙酯进行淬灭反应,再加入饱和食盐水洗涤,分出有机相,水相再用乙酸乙酯反复萃取3次合并有机相,加入无水硫酸钠干燥,经减压蒸馏除去溶剂,再经柱层析分离,分离后得到白色固体(72mg,分离产率78%)。
[0077]
反应完成后,白色固体经核磁共振(nuclear magnetic resonance spectroscopy,nmr)检测,氘代率为38%。
[0078]
实施例5
[0079]
本实施例提供了一种反式氘代二苯乙烯的合成方法,与实施例3的差异在于:硫源为硫化钠(39mg,0.5mmo1)。
[0080]
反应完成后,加入乙酸乙酯进行淬灭反应,再加入饱和食盐水洗涤,分出有机相,水相再用乙酸乙酯反复萃取3次合并有机相,加入无水硫酸钠干燥,经减压蒸馏除去溶剂,再经柱层析分离,分离后得到白色固体(80mg,分离产率87%)。
[0081]
反应完成后,白色固体经核磁共振(nuclear magnetic resonance spectroscopy,nmr)检测,氘代率为56%。
[0082]
实施例6
[0083]
本实施例提供了一种反式氘代二苯乙烯的合成方法,与实施例3的差异在于:硫源
为硫粉(16mg,0.5mmo1)。
[0084]
反应完成后,加入乙酸乙酯进行淬灭反应,再加入饱和食盐水洗涤,分出有机相,水相再用乙酸乙酯反复萃取3次合并有机相,加入无水硫酸钠干燥,经减压蒸馏除去溶剂,再经柱层析分离,分离后得到白色固体(78mg,分离产率85%)。
[0085]
反应完成后,白色固体经核磁共振(nuclear magnetic resonance spectroscopy,nmr)检测,氘代率为20%。
[0086]
实施例7
[0087]
本实施例提供了一种反式氘代二苯乙烯的合成方法,与实施例3的差异在于:硫源为乙基黄原酸钾(90mg,0.5mmo1)。
[0088]
反应完成后,加入乙酸乙酯进行淬灭反应,再加入饱和食盐水洗涤,分出有机相,水相再用乙酸乙酯反复萃取3次合并有机相,加入无水硫酸钠干燥,经减压蒸馏除去溶剂,再经柱层析分离,分离后得到白色固体(84mg,分离产率92%)。
[0089]
反应完成后,白色固体经核磁共振(nuclear magnetic resonance spectroscopy,nmr)检测,氘代率为92%。
[0090]
本实施例的白色固体表征数据如下:
[0091]1h nmr(500mhz,cdcl3)δ7.64
–
7.60(m,4h),7.46(t,j=7.6hz,4h),7.37(t,j=7.4hz,2h),7.22(s,0.20h).
[0092]
13
c nmr(125mhz,cdcl3)δ137.2(2c),128.6(4c),127.5(2c),126.4(4c).
[0093]2h nmr(77mhz,ch2cl2)δ7.22(s).
[0094]
hrms(esi
‑
tof)m/z[m+h]
+
:calcd for c
14
h
11
d
2 183.1137;found 183.1135.
[0095]
实施例8
[0096]
本实施例提供了一种反式氘代二苯乙烯的合成方法,与实施例7的差异在于:溶剂为n,n
‑
二甲基乙酰胺(2ml)。
[0097]
反应完成后,加入乙酸乙酯进行淬灭反应,再加入饱和食盐水洗涤,分出有机相,水相再用乙酸乙酯反复萃取3次合并有机相,加入无水硫酸钠干燥,经减压蒸馏除去溶剂,再经柱层析分离,分离后得到白色固体(84mg,分离产率92%)。
[0098]
反应完成后,白色固体经核磁共振(nuclear magnetic resonance spectroscopy,nmr)检测,氘代率为55%。
[0099]
实施例9
[0100]
本实施例提供了一种反式氘代二苯乙烯的合成方法,与实施例7的差异在于:溶剂为n
‑
甲基吡咯烷酮(2ml)。
[0101]
反应完成后,加入乙酸乙酯进行淬灭反应,再加入饱和食盐水洗涤,分出有机相,水相再用乙酸乙酯反复萃取3次合并有机相,加入无水硫酸钠干燥,经减压蒸馏除去溶剂,再经柱层析分离,分离后得到白色固体(82mg,分离产率90%)。
[0102]
反应完成后,白色固体经核磁共振(nuclear magnetic resonance spectroscopy,nmr)检测,氘代率为65%。
[0103]
实施例10
[0104]
本实施例提供了一种反式氘代二苯乙烯的合成方法,与实施例7的差异在于:溶剂为二甲苯(2ml)。
[0105]
反应完成后,加入乙酸乙酯进行淬灭反应,再加入饱和食盐水洗涤,分出有机相,水相再用乙酸乙酯反复萃取3次合并有机相,加入无水硫酸钠干燥,经减压蒸馏除去溶剂,再经柱层析分离,分离后得到白色固体(84mg,分离产率92%)。
[0106]
反应完成后,白色固体经核磁共振(nuclear magnetic resonance spectroscopy,nmr)检测,氘代率为25%。
[0107]
实施例11
[0108]
本实施例提供了一种反式氘代二苯乙烯的合成方法,与实施例7的差异在于:溶剂为二甲基亚砜(2ml)。
[0109]
反应完成后,加入乙酸乙酯进行淬灭反应,再加入饱和食盐水洗涤,分出有机相,水相再用乙酸乙酯反复萃取3次合并有机相,加入无水硫酸钠干燥,经减压蒸馏除去溶剂,再经柱层析分离,分离后得到白色固体(80mg,分离产率87%)。
[0110]
反应完成后,白色固体经核磁共振(nuclear magnetic resonance spectroscopy,nmr)检测,氘代率为30%。
[0111]
实施例12
[0112]
本实施例提供了一种反式氘代二苯乙烯的合成方法,与实施例7的差异在于:反应时间为12h。
[0113]
反应完成后,加入乙酸乙酯进行淬灭反应,再加入饱和食盐水洗涤,分出有机相,水相再用乙酸乙酯反复萃取3次合并有机相,加入无水硫酸钠干燥,经减压蒸馏除去溶剂,再经柱层析分离,分离后得到白色固体(83mg,分离产率90%)。
[0114]
反应完成后,白色固体经核磁共振(nuclear magnetic resonance spectroscopy,nmr)检测,氘代率为80%。
[0115]
实施例13
[0116]
本实施例提供了一种(e)
‑1‑
甲基
‑4‑
(2
‑
苯基乙烯基
‑
1,2
‑
d2)苯的合成方法,包括如下式所示的步骤:
[0117][0118]
分别加入(e)
‑1‑
甲基
‑4‑
苯乙烯基苯(97mg,0.5mmo1)、乙基黄原酸钾(80mg,0.5mmo1)和重水(0.5ml),在n,n
‑
二甲基乙酰胺(2ml)作溶剂的条件下,在反应温度为150℃情况下搅拌反应24小时,反应完成后,加入乙酸乙酯进行淬灭反应,再加入饱和食盐水洗涤,分出有机相,水相再用乙酸乙酯反复萃取3次合并有机相,加入无水硫酸钠干燥,经减压蒸馏除去溶剂,再经柱层析分离,分离后得到棕色固体(85mg,分离产率87%)。
[0119]
反应完成后,棕色固体经核磁共振(nuclear magnetic resonance spectroscopy,nmr)检测,氘代率为95%。
[0120]
本实施例棕色固体的表征数据如下:
[0121]
nmr(500mhz,cdcl3)δ7.53(d,j=7.9hz,2h),7.44(d,j=8.1hz,2h),7.38(t,j=7.7hz,2h),7.28(d,j=7.3hz,1h),7.20(d,j=7.8hz,2h),7.17(d,j=4.1hz,0.10h),2.39(s,3h).
[0122]
13
c nmr(125mhz,cdcl3)δ137.5(2c),134.4,129.4,128.6,128.6,127.6,127.4,126.5
–
126.3(m,2c),21.2.
[0123]2h nmr(77mhz,ch2cl2)δ7.15(s),7.10(s).
[0124]
hrms(esi
‑
tof)m/z[m+h]
+
:calcd for c
15
h
13
d
2 197.1294;found 197.1289.
[0125]
实施例14
[0126]
本实施例提供了一种(e)
‑1‑
甲基
‑2‑
(2
‑
苯基乙烯基
‑
1,2
‑
d2)苯的合成方法,合成路线包括如下式所示的步骤:
[0127][0128]
分别加入(e)
‑1‑
甲基
‑2‑
(2
‑
苯基乙烯基)苯(97mg,0.5mmo1)、乙基黄原酸钾(80mg,0.5mmo1)和重水(0.5ml),在n,n
‑
二甲基乙酰胺(2ml)作溶剂的条件下,在反应温度为150℃情况下搅拌反应24小时,反应完成后,加入乙酸乙酯进行淬灭反应,再加入饱和食盐水洗涤,分出有机相,水相再用乙酸乙酯反复萃取3次合并有机相,加入无水硫酸钠干燥,经减压蒸馏除去溶剂,再经柱层析分离,分离后得到黄色固体(83mg,分离产率85%)。
[0129]
反应完成后,黄色固体经核磁共振(nuclear magnetic resonance spectroscopy,nmr)检测,氘代率为99%。
[0130]
本实施例黄色固体的表征数据如下:
[0131]1h nmr(500mhz,cdcl3)δ7.63(d,j=7.4hz,1h),7.55(d,j=7..2hz,2h),7.39(t,j=7.6hz,2h),7.29(t,j=7.4hz,1h),7.25
–
7.22(m,1h),7.21(d,j=4.0hz,2h),2.46(s,3h).
[0132]
13
c nmr(125mhz,cdcl3)δ137.5,136.2,135.8,130.4,128.7(2c),127.6,127.5,126.5(2c),126.2,125.3,19.9.
[0133]2h nmr(77mhz,ch2cl2)δ7.19(s),6.84(s).
[0134]
hrms(esi
‑
tof)m/z[m+h]
+
:calcd for c
15
h
13
d
2 197.1294;found 197.1287.
[0135]
实施例15
[0136]
本实施例提供了一种(e)
‑
1,2
‑
二甲基
‑4‑
(2
‑
苯基乙烯基
‑
1,,2
‑
d2)苯的合成方法,合成路线包括如下式所示的步骤:
[0137][0138]
分别加入(e)
‑
1,2
‑
二甲基
‑4‑
(2
‑
苯基乙烯基)苯(104mg,0.5mmo1)、乙基黄原酸钾(80mg,0.5mmo1)和重水(0.5ml),在n,n
‑
二甲基乙酰胺(2ml)作溶剂的条件下,在反应温度为150℃情况下搅拌反应24小时,反应完成后,加入乙酸乙酯进行淬灭反应,再加入饱和食盐水洗涤,分出有机相,水相再用乙酸乙酯反复萃取3次合并有机相,加入无水硫酸钠干燥,经减压蒸馏除去溶剂,再经柱层析分离,分离后得到白色固体(92mg,分离产率88%)。
[0139]
反应完成后,白色固体经核磁共振(nuclear magnetic resonance spectroscopy,nmr)检测,氘代率为96%。
[0140]
本实施例白色固体的表征数据如下:
[0141]1h nmr(500mhz,cdcl3)δ7.52(d,j=7.3hz,2h),7.37(t,j=7.7hz,2h),7.33(s,1h),7.27(q,j=7.1,6.3hz,2h),7.14(d,j=7.7hz,1h),7.09(s,0.08h),,2.31(s,3h),2.29(s,3h).
[0142]
13
c nmr(125mhz,cdcl3)δ137.5,136.8,136.2,134.8,129.9,128.6(2c),127.7,127.3,126.3(2c),124.0,19.8,19.6.
[0143]2h nmr(77mhz,ch2cl2)δ7.09(s).
[0144]
hrms(esi
‑
tof)m/z[m+h]
+
:calcd for c
16
h
15
d
2 211.1450;found 211.1448.
[0145]
实施例16
[0146]
本实施例提供了一种(e)
‑1‑
甲氧基
‑4‑
(2
‑
苯基乙烯基
‑
1,2
‑
d2)苯的合成方法,合成路线包括如下式所示的步骤:
[0147][0148]
分别加入(e)
‑1‑
甲氧基
‑4‑
(2
‑
苯基乙烯基)苯(105mg,0.5mmo1)、乙基黄原酸钾(80mg,0.5mmo1)和重水(0.5ml),在n,n
‑
二甲基乙酰胺(2ml)作溶剂的条件下,在反应温度为150℃情况下搅拌反应24小时,反应完成后,加入乙酸乙酯进行淬灭反应,再加入饱和食盐水洗涤,分出有机相,水相再用乙酸乙酯反复萃取3次合并有机相,加入无水硫酸钠干燥,经减压蒸馏除去溶剂,再经柱层析分离,分离后得到白色固体(85mg,分离产率80%)。
[0149]
反应完成后,白色固体经核磁共振(nuclear magnetic resonance spectroscopy,nmr)检测,氘代率为80%。
[0150]
本实施例白色固体的表征数据如下:
[0151]1h nmr(500mhz,cdcl3)δ7.48(dd,j=16.7,8.0hz,4h),7.35(t,,j=7.6hz,2h),7.23(d,j=7.4hz,1h),6.91(d,j=8.7hz,2h),7.10
–
6.96(m,0.40h),3.84(s,3h).
[0152]
13
c nmr(125mhz,cdcl3)δ159.2,137.5,130.0,128.6(2c),127.7(2c),127.2,126.2(2c),114.1(2c),55.30.
[0153]2h nmr(77mhz,ch2cl2)δ7.04(s).
[0154]
hrms(esi
‑
tof)m/z[m+h]
+
:calcd for c
15
h
13
d2o 213.1243;found 213.1242.
[0155]
实施例17
[0156]
本实施例提供了一种(e)
‑4‑
(2
‑
苯基乙烯基
‑
1,2
‑
d2)苯酚的合成方法合成路线包括如下式所示的步骤:
[0157][0158]
分别加入(e)
‑4‑
(2
‑
苯基乙烯基)苯酚(98mg,0.5mmo1)、乙基黄原酸钾(80mg,
0.5mmo1)和重水(0.5ml),在n,n
‑
二甲基乙酰胺(2ml)作溶剂的条件下,在反应温度为150℃情况下搅拌反应24小时,反应完成后,加入乙酸乙酯进行淬灭反应,再加入饱和食盐水洗涤,分出有机相,水相再用乙酸乙酯反复萃取3次合并有机相,加入无水硫酸钠干燥,经减压蒸馏除去溶剂,再经柱层析分离,分离后得到白色固体(89mg,分离产率90%)。
[0159]
反应完成后,白色固体经核磁共振(nuclear magnetic resonance spectroscopy,nmr)检测,氘代率为90%。
[0160]
本实施例白色固体的表征数据如下:
[0161]1h nmr(500mhz,acetone
‑
d6)δ8.52(s,1h),7.55(d,j=7.6hz,2h),7.46(s,2h),7.34(t,j=7.7hz,2h),7.22(t,j=7.4hz,1h),7.17(d,j=16..4hz,1h),7.05(d,j=16.4hz,1h),6.85(d,j=9.0hz,0.19h).
[0162]
13
c nmr(125mhz,acetone
‑
d6)δ157.3,138.0,128.6(2c),128.5,127.8(2c),127.0(2c),126.1(2c),125.6.
[0163]2h nmr(77mhz,ch2cl2)δ6.83(s,1h).
[0164]
hrms(esi
‑
tof)m/z[m+h]
+
:calcd for c
14
h
11
d2o 199.1086;found 199.1084.
[0165]
实施例18
[0166]
本实施例提供了一种(e)
‑1‑
氟
‑4‑
(2
‑
苯基乙烯基
‑
1,2
‑
d2)苯的合成方法,合成路线包括如下式所示的步骤:
[0167][0168]
分别加入(e)
‑1‑
氟
‑4‑
(2
‑
苯基乙烯基)苯(99mg,0.5mmo1)、乙基黄原酸钾(80mg,0.5mmo1)和重水(0.5ml),在n,n
‑
二甲基乙酰胺(2ml)作溶剂条件下,在反应温度为150℃情况下搅拌反应24小时,反应完成后,加入乙酸乙酯进行淬灭反应,再加入饱和食盐水洗涤,分出有机相,水相再用乙酸乙酯反复萃取3次合并有机相,加入无水硫酸钠干燥,经减压蒸馏除去溶剂,再经柱层析分离,分离后得到白色固体(85mg,分离产率85%)。
[0169]
反应完成后,白色固体经核磁共振(nuclear magnetic resonance spectroscopy,nmr)检测,氘代率为99%。
[0170]
本实施例白色固体的表征数据如下:
[0171]1h nmr(500mhz,cdcl3)δ7.56
–
7.46(m,4h),7.38(t,j=7.6hz,2h),7.28(t,j=7.4hz,1h),7.07(t,j=8.7hz,2h).
[0172]
13
c nmr(125mhz,cdcl3)δ162.3(d,j=247.1hz),137.0,133.4,128.7(2c),127.9(d,j=8.0hz,2c),127.6(2c),126.4(2c),115.6(d,j=21.6hz).
[0173]2h nmr(77mhz,ch2cl2)δ7.06(s),7.01(s).
[0174]
hrms(esi
‑
tof)m/z[m+h]
+
:calcd for c
14
h
10
d2f 201.1043;found 201.1045.
[0175]
实施例19
[0176]
本实施例提供了一种(e)
‑1‑
氯
‑4‑
(2
‑
苯基乙烯基
‑
1,2
‑
d2)苯的合成方法,合成路线包括如下式所示的步骤:
[0177][0178]
分别加入(e)
‑1‑
氯
‑4‑
(2
‑
苯基乙烯基)(107mg,0.5mmo1)、乙基黄原酸钾(80mg,0.5mmo1)和重水(0.5ml),在n,n
‑
二甲基乙酰胺(2ml)条件下,反应温度为150℃情况下搅拌反应24小时反应完成后,加入乙酸乙酯进行淬灭反应,再加入饱和食盐水洗涤,分出有机相,水相再用乙酸乙酯反复萃取3次合并有机相,加入无水硫酸钠干燥,经减压蒸馏除去溶剂,再经柱层析分离,分离后得到白色固体(89mg,,分离产率83%)。
[0179]
反应完成后,白色固体经核磁共振(nuclear magnetic resonance spectroscopy,nmr)检测,氘代率为98%。
[0180]
本实施例白色固体的表征数据如下:
[0181]1h nmr(500mhz,cdcl3)δ7.53(d,j=7.8hz,2h),7.45(d,j=7..5hz,2h),7.40(t,j=7.5hz,2h),7.35(d,j=8.4hz,2h),7.31(t,j=7.3hz,1h),7.09(s,0.2h).
[0182]
13
c nmr(125mhz,cdcl3)δ136.8,135.7,133.1,128.8(2c),128.7(2c),127.8,127.6(2c),126.5(2c).
[0183]2h nmr(77mhz,ch2cl2)δ7.06(s).
[0184]
hrms(esi
‑
tof)m/z[m+h]
+
:calcd for c
12
h
10
d2cl 217.0748;found 217.0745.
[0185]
实施例20
[0186]
本实施例提供了一种(e)
‑1‑
溴
‑4‑
(2
‑
苯基乙烯基
‑
1,2
‑
d2)苯的合成方法,合成路线包括如下式所示的步骤:
[0187][0188]
分别加入(e)
‑1‑
溴
‑4‑
(2
‑
苯基乙烯基)苯(129mg,0.5mmo1)、乙基黄原酸钾(80mg,0.5mmo1)和重水(0.5ml),在n,n
‑
二甲基乙酰胺(2ml)作溶剂的条件下,在反应温度为150℃情况下搅拌反应24小时,反应完成后,加入乙酸乙酯进行淬灭反应,再加入饱和食盐水洗涤,分出有机相,水相再用乙酸乙酯反复萃取3次合并有机相,加入无水硫酸钠干燥,经减压蒸馏除去溶剂,再经柱层析分离,分离后得到白色固体(110mg,分离产率85%)。
[0189]
反应完成后,白色固体经核磁共振(nuclear magnetic resonance spectroscopy,nmr)检测,氘代率为82%。
[0190]
本实施例白色固体的表征数据如下:
[0191]1h nmr(500mhz,cdcl3)δ7.50(dd,j=14.3,7.9hz,4h),7.38(dd,j=8.1,6.4hz,4h),7.29(t,j=7.3hz,1h),7.07(q,j=16.3hz,0.36h).
[0192]
13
c nmr(125mhz,cdcl3)δ136.81,136.14,131.7(2c),128.7(2c),127.9(2c),127.86,126.5(2c),121.25.
[0193]2h nmr(77mhz,ch2cl2)δ7.07(s).
[0194]
hrms(esi
‑
tof)m/z[m+h]
+
:calcd for c
14
h
10
d2br 261.0243;found 261.0251.
[0195]
实施例21
[0196]
本实施例提供了一种(e)
‑3‑
(2
‑
苯基乙烯基
‑
1,2
‑
d2)噻吩的合成方法,,合成路线包括如下式所示的步骤:
[0197][0198]
分别加入(e)
‑3‑
(2
‑
苯基乙烯基)噻吩(93mg,0.5mmo1)、乙基黄原酸钾(80mg,0.5mmo1)、重水(0.5ml),在n,n
‑
二甲基乙酰胺(2ml)作溶剂的条件下,在反应温度为150℃情况下搅拌反应24小时,反应完成后,加入乙酸乙酯进行淬灭反应,再加入饱和食盐水洗涤,分出有机相,水相再用乙酸乙酯反复萃取3次合并有机相,加入无水硫酸钠干燥,经减压蒸馏除去溶剂,再经柱层析分离,分离后得到白色固体(82mg,分离产率87%)。
[0199]
反应完成后,白色固体经核磁共振(nuclear magnetic resonance spectroscopy,nmr)检测,氘代率为80%。
[0200]
本实施例白色固体的表征数据如下:
[0201]1h nmr(500mhz,cdcl3)δ7.51(d,j=7.5hz,2h),7.39(t,j=7.6hz,3h),7.35(t,j=4.2hz,1h),7.28(dd,j=14.1,6.7hz,2h),7.17(d,j=16.3hz,0.48h),6.99(d,j=16.3hz,0.48h).
[0202]
13
c nmr(125mhz,cdcl3)δ140.0,137.3,128.6(2c),127.4,126.2(2c),124.9,122.8,122.3.
[0203]2h nmr(77mhz,ch2cl2)δ7.39
–
6.79(m).
[0204]
hrms(esi
‑
tof)m/z[m+h]
+
:calcd for c
12
h9d2s 189.0701;found 189.0698.
[0205]
实施例22
[0206]
本实施例提供了一种1,4
‑
二(4
‑
吡啶基
‑
1,2
‑
d2)乙烯的合成方法,合成路线包括如下式所示的步骤:
[0207][0208]
分别加入1,4
‑
二(4
‑
吡啶基)乙烯(110mg,0.5mmo1)、乙基黄原酸钾(80mg,0.5mmo1)和重水(0.5ml),在n,n
‑
二甲基乙酰胺(2ml)作溶剂的条件下,在反应温度为150℃情况下搅拌反应24小时,反应完成后,加入乙酸乙酯进行淬灭反应,再加入饱和食盐水洗涤,分出有机相,水相再用乙酸乙酯反复萃取3次合并有机相,加入无水硫酸钠干燥,经减压蒸馏除去溶剂,再经柱层析分离,分离后得到白色固体(77mg,分离产率84%)。
[0209]
反应完成后,白色固体经核磁共振(nuclear magnetic resonance spectroscopy,nmr)检测,氘代率为99%。
[0210]
本实施例白色固体的表征数据如下:
[0211]1h nmr(500mhz,cdcl3)δ8.48(d,j=5.9hz,4h),7.05(d,j=5..9hz,4h).
[0212]
13
c nmr(125mhz,cdcl3)δ149.8(4c),149.3(2c),123.8(4c).
[0213]2h nmr(77mhz,ch2cl2)δ6.90(s).
[0214]
hrms(esi
‑
tof)m/z[m+k]
+
:calcd for c
12
h8d2nk 223.0601;found 223.0591.
[0215]
本发明实施方式中的合成方法为反式氘代二苯乙烯提供了一种简洁高效的制备方法。对同类产品及下游产品的工艺开发具有良好的借鉴意义。
[0216][0217]
本发明部分实施方式中合成方法的反应机理如上式所示:etocs2k在加热条件下分解产生的三硫自由基阴离子(s3·
‑
)对烯烃经自由基加成得到烷基自由基中间体a。接着,在(s3·
‑
)的帮助下,环硫过渡态b可以形成并立刻从etocs2d(来源于引发剂etocs2k和过量d2o)获得氘原子得到单氘代1,2,3
‑
三硫戊环c。然后,1,2,3
‑
三硫烷c在脱硫反应下生环硫d。环硫d同样在(z
‑
s
‑
)的作用下通过脱硫反应生成单硫代烯烃e。在这个过程中,(z
‑
s
‑
)代表任何硫介质中的阴离子(黄原酸盐和硫化物等)。单硫代烯烃e重复上述环化和脱硫反应可以得到双氘代的环硫g,经过脱硫反应生成顺式二代烯烃。顺式烯烃在热力学条件下转化为反式烯烃2。在单氘取代烯烃e转化为双氘代1,2,3
‑
三硫烷f的时候,由于c
‑
d键的键能远大于c
‑
h键,所以反应过程总是优先脱除氢原子而保留氘原子。
[0218]
综上所述,本发明以烯烃化合物为原料,无过渡金属催化,在添加引发剂的条件下,加热搅拌反应;通过一锅法合成了反式氘代烯烃化合物,除终产物外,无需针对转化过程中的中间体(相关技术中选用的过渡金属催化剂和配体及其中间产物)进行分离和纯化,一步完成合成,容易实现产业化生产,为工业生产中减少了资金和劳动力投入。
[0219]
上面结合具体实施方式对本发明实施例作了详细说明,但是本发明不限于上述实施例,在所属技术领域普通技术人员所具备的知识范围内,还可以在不脱离本发明宗旨的前提下作出各种变化。此外,在不冲突的情况下,本发明的实施例及实施例中的特征可以相互组合。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1