酒石酸伐尼克兰的相关杂质化合物及其制备、应用和检测方法与流程
1.本发明属于药物领域,具体涉及酒石酸伐尼克兰的相关杂质化合物及其制备、应用和检测方法。
背景技术:
2.伐尼克兰,是一种芳基稠合氮杂多环化合物,能够调节胆碱能,以酒石酸盐的形式出售,商品名为或能与神经元烟碱乙酰胆碱特异性受体位点结合,可用于以下疾病:
3.治疗炎性肠疾病(包括但不限于:溃疡性结肠炎、坏疽性脓皮病和克罗恩氏病(crohn’s disease))、过敏性肠综合症、痉挛性张力障碍、慢性疼痛、急性疼痛、腹泻(celiac sprue)、囊炎(pouchitis)、血管收缩、焦虑、恐慌症(panic disorder)、抑郁症、双相性精神障碍(bipolar disorder)、孤独症、睡眠障碍、时差反应(jet lag)、肌萎缩性侧索硬化(als)、认知机能障碍、药物/毒素-诱发的认知损伤(例如,起因于酒精、巴比妥类药物、维生素缺乏、消遣性药物(recreational drug)、铅、砷、汞)、疾病诱发的认知损伤(例如,起因于阿尔茨海默氏病(早老性痴呆)、血管性痴呆(vascular dementia)、帕金森氏病、多发性硬化症、aids、(大)脑炎、创伤、肾性脑病和肝性脑病、甲状腺机能减退、皮克氏病、科尔萨科夫氏综合症和前额(frontal)和皮层下痴呆(subcortical dementia))、高血压、食欲过盛、厌食症、肥胖症、心律失常、胃酸分泌过多、溃疡、嗜铬细胞瘤、进行性核上麻痹(progressive supramuscular palsy)、化学品依赖和成瘾(例如,对尼古丁(和/或烟草产品)、酒精、苯并二氮类、巴比妥类、类阿片或可卡因的依赖和成瘾)、头痛、偏头痛、中风、外伤性脑损伤(tbi)、强迫观念与行为性疾病(ocd)、精神病、亨廷顿氏舞蹈病、迟发性运动障碍、运动机能亢进、诵读困难、精神分裂症、多发性硬化性痴呆、年龄相关性认知衰退、癫痫症、包括小发作缺乏癫痫症(including petit mal absence epilepsy)、注意力缺陷机能亢进障碍(attention deficit hyperactivity disorder)(adhd)、图雷特氏综合症(tourette’s syndrome);特别是,对尼古丁的依赖、成瘾和脱瘾,包括在戒烟治疗中的应用(参见:cn 1509174a)。
4.伐尼克兰的制备方法,以及其消旋物的拆分方法,已被美国专利申请us 6,410,550和国际专利申请wo 01/62736所公开,它们的内容也可以通过引用的方式并入本文。
5.亚硝胺化合物,包括与胺结合的亚硝基(n(r1)(r2)-n=o)结构,是某些动物的基因毒性物质,有些已被国际癌症研究机构(iarc)列为可能或潜在的人类致癌物。
6.这些亚硝胺化合物,在人用药物注册技术要求国际协调会(international conference on harmonization,简称ich)指导文件m7(r1)中被称为“关注组群”化合物,评估和控制药品中的dna反应性(致突变)杂质以限制潜在的致癌风险(2018年3月)。ich指南建议,将包括亚硝胺化合物在内的任何已知致突变/致癌物控制在人类癌症风险可忽略的摄入量水平。
7.目前,美国食品和药物管理局(fda)已鉴定出七种亚硝胺杂质,这些杂质理论上可能存在于药品中,因为使用了可能导致亚硝胺形成的制备工艺和材料:n,n-二甲基亚硝胺(ndma)、n-亚硝基二乙胺(ndea)、n-亚硝基-n-甲基-4-氨基丁酸(nmba)、n亚硝基异丙基乙胺(nipea)、n-亚硝二异丙胺(ndipa)、n亚硝二丁胺(ndba)和n-亚硝基甲基苯胺(nmpa)。其中,有五种杂质(ndma、ndea、nmba、nipea和nmpa)实际上已在原料药或药品中检测到。
8.举例来说,fda试验的一些初步结果表明,某些雷尼替丁产品中的ndma含量已超过可接受的水平。近年来,fda还发现有的二甲双胍产品中含有ndma杂质,且在某些批次中检出ndma含量高于fda建议的可接受摄入量限值。
9.有鉴于此,特提出本发明,以便将酒石酸伐尼克兰原料药或制剂中的亚硝胺杂质含量控制在能够满足安全性要求的较低水平。
技术实现要素:
10.针对现有技术存在的问题和/或不足,本发明的目的之一是提供一种与酒石酸伐尼克兰相关的新化合物:式ⅱ所示化合物。该新化合物会随着反应进程转变成具有基因毒性的亚硝胺杂质p08,需要在酒石酸伐尼克兰合成的全过程中加以检测和控制,以便进一步采取积极有效的技术手段,将酒石酸伐尼克兰原料药或制剂中的亚硝胺基因毒性杂质含量控制在较低水平,符合安全性要求。
11.本发明提供了一种式ⅱ所示化合物:
[0012][0013]
其中,r
11
、r
12
独立地选自硝基(-no2)或氨基(-nh2)。
[0014]
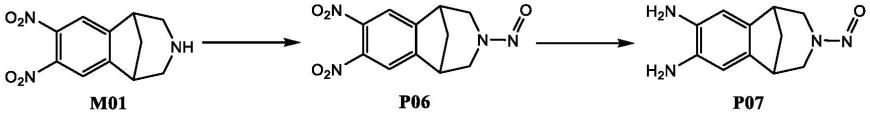
本发明还提供了一种式ⅱ所示化合物的制备方法,包括以下步骤:化合物m01与亚硝酸盐在酸和混合溶剂存在的条件下发生亚硝胺化反应,生成化合物p06;之后可选择地包括:化合物p06与还原剂在催化剂和卤代烃类溶剂存在的条件下发生还原反应,生成化合物p07;
[0015][0016]
所述的式ⅱ所示化合物为化合物p06或化合物p07;
[0017]
优选地,
[0018]
所述的亚硝酸盐为亚硝酸钾或亚硝酸钠;
[0019]
所述的酸为醋酸;
[0020]
所述的混合溶剂为水与四氢呋喃,所述水与四氢呋喃的体积比为1:4~6;
[0021]
所述的还原剂为氢气;
[0022]
所述的催化剂为钯炭催化剂;
[0023]
所述卤代烃类溶剂为液态的卤素取代的c1~c4烷烃,再优选为二氯甲烷;
[0024]
更优选地,
reaction monitoring)或mrm(multi reaction monitoring,多反应检测扫描),优选sim或mrm;
[0051]
优选地,
[0052]
所述液相色谱-串联质谱法的质谱条件还包括:离子源温度:350℃;毛细管电压:6000v:喷雾气流速:13l/min。
[0053]
在本发明的某一方案中,所述的式ⅱ所示化合物为(p06)时,流动相a与流动相b的体积比为55%:45%~75%:25%,所述的流动相a为甲酸-纯化水,甲酸与纯化水的体积比为0.05~0.2:100(例如,0.06:100、0.07:100、0.08:100、0.09:100、0.1:100、0.11:100、0.12:100、0.15:100等);优选地,流动相a与流动相b的体积比为63%:37%,甲酸与纯化水的体积比为0.1:100。
[0054]
在本发明的某一方案中,所述的式ⅱ所示化合物为(p07)时,流动相a与流动相b的体积比为90%:10%~98%:2%,所述的流动相a为乙酸铵水溶液,所述乙酸铵水溶液的浓度为0.005~0.02mol/l(例如,0.006mol/l、0.007mol/l、0.008mol/l、0.009mol/l、0.01mol/l、0.011mol/l、0.012mol/l、0.015mol/l等),用甲酸调节ph=2~3;优选地,流动相a与流动相b的体积比为95%:5%,所述乙酸铵水溶液的浓度为0.01mol/l,用甲酸调节ph=2.5。
[0055]
在本发明的某一方案中,所述的检测方法为定性检测或定量检测,标准品、对照品和酒石酸伐尼克兰的溶剂为水或水-乙腈,水与乙腈的体积比为90%:10%~98%:2%,优选95%:5%。
[0056]
在本发明的某一方案中,采用内标法或外标法进行定量检测;
[0057]
优选地,
[0058]
所述的外标法是:以式ⅱ所示化合物作为标准品或对照品,制成不同浓度的标准品溶液或对照品溶液,进样检测,得到对应的峰面积数据,将浓度与峰面积进行线性回归,得到线性方程;再将酒石酸伐尼克兰检测样品溶液中式ⅱ所示化合物的峰面积检测数据代入线性方程,可得到检测样品溶液中式ⅱ所示化合物的浓度数据;然后再用该数据除以检测样品溶液的浓度数据,即得定量检测结果。
[0059]
本发明的有益效果主要在于:本发明提供的式ⅱ所示化合物,会随着反应进程转变成具有基因毒性的亚硝胺杂质p08,以其作为标准品、对照品或检测项目用于酒石酸伐尼克兰或其原料、中间体的杂质研究、质量控制等,实现该杂质的定性和/或定量检测,以便进一步采取积极有效的技术手段,将酒石酸伐尼克兰原料药或制剂中的亚硝胺基因毒性杂质含量控制在较低的水平,从而符合安全性要求和药物产品监管规定,更好地保障患者用药安全。
附图说明
[0060]
图1为高效液相色谱法检测化合物a中亚硝胺杂质化合物p06的色谱图。
具体实施方式
[0061]
下面将结合具体实施例对本发明进行清楚、完整的描述,本领域技术人员将会理解,下面所述的实施例是本发明一部分实施例,而不是全部的实施例,仅用于说明本发明,而不应视为对本发明保护范围的限制。
[0062]
本发明中,未注明具体条件者,按照常规条件或制造商建议的条件进行,所用试剂或仪器未注明生产厂商者,均为可以通过市售购买获得的常规产品。
[0063]
关于本发明中使用术语的定义,除非另有说明,本文中术语提供的初始定义适用于全文中的该术语;对于本文没有具体定义的术语,应当根据公开内容和/或上下文,给出本领域技术人员能够给予它们的含义。
[0064]
实施例1
[0065]
亚硝胺化合物p06的制备
[0066][0067]
向100ml三口瓶中,加入5g化合物m01和52ml四氢呋喃(thf),搅拌溶清;加入10ml的nano2水溶液(浓度为0.4g/ml),以及3g醋酸,升温至50℃反应,tlc或hplc监控反应完毕,浓缩,加入50ml二氯甲烷(dcm)和50ml饱和食盐水,搅拌溶清,静置,分液,取有机相,用饱和食盐水洗一次,无水硫酸钠干燥,过滤,浓缩至干,得到亚硝胺化合物p06,纯度为93.69%(面积归一化法)。
[0068][0069]
亚硝胺化合物p06的1h-nmr、
13
c-nmr和ir图谱数据,分别见表1~3;m+h
+
=279.10;uv:在乙腈溶液中最大吸收波长为223.80nm。
[0070]
表1、亚硝胺化合物p06的1h-nmr图谱
[0071]
[0072][0073]
表2、亚硝胺化合物p06的
13
c-nmr图谱
[0074][0075]
表3、亚硝胺化合物p06的ir图谱
[0076]
吸收波数(cm-1
)归属3034.5ar-h2920.2,2879.0饱和c-h伸缩振动1667.2苯环c=c伸缩振动1546.9,1428.2,1344.9n=o,n-o伸缩振动845.2苯环c-h弯曲振动
[0077]
实施例2
[0078]
亚硝胺化合物p07的制备
[0079][0080]
向100ml三口瓶中,加入4.0g化合物p06和50ml二氯甲烷(dcm),搅拌溶清;加入1.0g钯炭催化剂(pd/c),先用氮气置换后,然后再持续通氢气,升温至25~30℃,反应过夜,tlc监控反应完毕,过滤,浓缩至干,得到亚硝胺化合物p07,纯度为96.59%(面积归一化法)。
[0081]
[0082]
亚硝胺化合物p07的1h-nmr、
13
c-nmr和ir图谱数据,分别见表4~6;m+h
+
=219.20;uv:在乙腈:水=1:1溶液中最大吸收波长为212.00nm。
[0083]
表4、亚硝胺化合物p07的1h-nmr图谱
[0084][0085]
表5、亚硝胺化合物p07的
13
c-nmr图谱
[0086][0087]
表6、亚硝胺化合物p07的ir图谱
[0088][0089][0090]
实施例3
[0091]
亚硝胺化合物p08的制备
[0092][0093]
向100ml三口瓶中,加入4.0g化合物m02(伐尼克兰)和45ml四氢呋喃(thf),搅拌溶清;加入8.2ml的nano2水溶液(浓度为0.4g/ml),以及2.9g醋酸,升温至52℃,tlc监控反应完毕,有固体析出,过滤,真空干燥,得到亚硝胺化合物p08,其纯度为98.87%(面积归一化法)。
[0094][0095]
亚硝胺化合物p08的1h-nmr、
13
c-nmr和ir图谱数据,分别见表7~9;m+h
+
=241.20;uv:在乙腈溶液中最大吸收波长为204.40nm。
[0096]
表7、亚硝胺化合物p08的1h-nmr图谱
[0097][0098]
表8、亚硝胺化合物p08的
13
c-nmr图谱
[0099]
[0100][0101]
表9、亚硝胺化合物p08的ir图谱
[0102]
吸收波数(cm-1
)归属3048.8ar-h2961.4,2931.8饱和c-h伸缩振动1925.2苯环c=c伸缩振动1575.2n=o伸缩振动885.2苯环c-h弯曲振动
[0103]
实施例4
[0104]
申请日为2020年07月20日的中国专利申请202010698570.8(发明名称:一种伐尼克兰中间体、伐尼克兰及其盐的制备方法),其全部内容以引用方式全部并入本文。
[0105]
1、伐尼克兰中间体的制备
[0106][0107]
①
、将3kg(约8.69mol)化合物a和0.35kg钯炭(pd/c,pd 5%)催化剂,加入到36kg异丙醇和15kg纯化水的反应釜中,控制反应温度在25~35℃,通氢气并搅拌进行反应6h,然后停止反应(hplc检测反应液中化合物a的剩余量≤0.5%),垫硅藻土过滤除去钯炭催化剂,得到含化合物b的反应液;
[0108][0109]
②
、在惰性气体(本实施例为氮气)保护下,向步骤
①
所得的反应液中,加入75g碳酸氢钠(约0.89mol,显碱性),然后缓慢加入乙二醛的水溶液(0.556kg(约9.58mol)乙二醛和6.5kg水,显酸性)中,控制反应温度在20~30℃,搅拌反应8h(包含乙二醛水溶液的加入时间),然后停止反应(hplc检测反应液中化合物b的剩余量≤0.5%),减压蒸馏(真空度≤-0.09mpa,40~50℃)至基本无馏分产生,加入90kg纯化水,控制釜内温度在20~30℃,保温2h,之后离心,干燥(45~50℃),得到2.34kg伐尼克兰中间体(化合物c),为类白色固体,hplc纯度99.1%,有关物质检测项目:化合物a≤0.1%,化合物b≤0.1%,其他最大单个未
知杂质含量≤0.1%。
[0110]
2、酒石酸伐尼克兰的制备
[0111][0112]
a、在惰性气体(本实施例为氮气)保护下,向反应釜中加入20kg纯化水、0.81kg(约20.25mol)氢氧化钠、8kg二氯甲烷和2kg(约6.51mol)化合物c(前一步骤制得),控制反应温度在20~35℃,搅拌反应8h,然后停止反应(hplc检测反应液中化合物c的剩余量≤0.5%),之后向反应釜内加入18.5kg二氯甲烷,搅拌,静置,分液,取有机相,水相用二氯甲烷(15kg
×
2)萃取,合并有机相,有机相中加入5kg无水硫酸钠干燥,过滤,对滤液减压蒸馏(真空度≤-0.09mpa,40~50℃)至基本无馏分产生,加入10kg无水乙醇,继续减压蒸馏(真空度≤-0.09mpa,40~50℃)至基本无馏分产生,得到含伐尼克兰的母液;
[0113][0114]
b、向步骤a所得含伐尼克兰的母液中,缓慢加入30kg无水乙醇和1.16kg(约7.73mol)l-(+)-酒石酸,控制反应温度在20~30℃,搅拌反应16h(包含无水乙醇和l-(+)-酒石酸的加入时间),然后停止反应,之后离心,干燥(70~75℃),得到2.08kg酒石酸伐尼克兰,为类白色固体。
[0115]
实施例5
[0116]
根据fda(food and drug administration,是指美国食品药品监督管理局)要求,需要对药物中的亚硝胺杂质进行检测,一般要求控制在≤7.5ppm。
[0117]
为此,我们创造性开发了超高效液相色谱三重四极杆质谱联用法(uplc-ms/ms),用于本发明酒石酸伐尼克兰原料药中亚硝胺杂质(化合物p08)的定性和/或定量检测。uplc-ms/ms的色谱条件以及质谱条件,具体见表10。
[0118]
表10、uplc-ms/ms的色谱条件以及质谱条件
[0119]
[0120][0121]
经检测,实施例4制备的酒石酸伐尼克兰中亚硝胺杂质化合物p08的含量为114ppm(试验结果显示,该杂质的保留时间约在3.47min左右);这表明,如果不加以控制和处理,而按照常规的方法制备酒石酸伐尼克兰,所得产物中的亚硝胺杂质化合物p08含量通常在数百ppm级别,无法满足fda对亚硝胺杂质的限量要求。
[0122]
实施例6
[0123]
1、伐尼克兰中间体的制备
[0124]
①
、将3kg化合物a(经检测,化合物a中亚硝胺杂质化合物p06的含量为722ppm)和0.35kg钯炭(pd/c,pd 5%)催化剂,加入到36kg异丙醇和15kg纯化水的反应釜中,控制反应温度在25~35℃,通氢气并搅拌进行反应,hplc检测反应液中化合物a的剩余量≤0.5%停止反应,垫硅藻土过滤除去钯炭催化剂,得到含化合物b的反应液;
[0125]
②
、在惰性气体(氮气)保护下,向步骤
①
所得的反应液中,加入75g碳酸氢钠,然后缓慢加入乙二醛的水溶液(0.56kg乙二醛和6.5kg水)中,控制反应温度在20~30℃,搅拌反应,hplc检测反应液中化合物b的剩余量≤0.5%停止反应,减压蒸馏(真空度≤-0.09mpa,40~50℃)至基本无馏分产生,加入90kg纯化水,控制釜内温度在20~30℃,保温2h,之后离心,干燥(45~50℃),得到化合物c。
[0126]
2、酒石酸伐尼克兰的制备
[0127]
a、在惰性气体(氮气)保护下,向反应瓶中加入20kg纯化水、0.81kg氢氧化钠、8kg二氯甲烷和2kg化合物c(前一步骤制得),控制反应温度在20~35℃,hplc检测反应液中化
合物c的剩余量≤0.5%停止反应,之后向反应釜内加入18.5kg二氯甲烷,搅拌,静置,分液,取有机相,水相用二氯甲烷(15kg
×
2)萃取,合并有机相,有机相中加入5kg无水硫酸钠干燥,过滤,对滤液减压蒸馏(真空度≤-0.09mpa,40~50℃)至基本无馏分产生,加入10kg无水乙醇,继续减压蒸馏(真空度≤-0.09mpa,40~50℃)至基本无馏分产生,得到含伐尼克兰的母液;
[0128]
b、向步骤a所得含伐尼克兰的母液中,缓慢加入30kg无水乙醇和1.16kg的l-(+)-酒石酸,控制反应温度在20~30℃,搅拌反应16h,然后停止反应,之后离心,干燥(70~75℃),得到酒石酸伐尼克兰;
[0129]
再按照每克酒石酸伐尼克兰的溶剂用量8ml,将制得的酒石酸伐尼克兰加入到打浆溶剂中,于30~40℃打浆1.5h,之后离心,干燥,即可;经检测,二氯甲烷打浆处理后的酒石酸伐尼克兰中,亚硝胺杂质化合物p08的含量为5.94ppm,亚硝胺杂质化合物p06和p07的含量为未检出(小于检测限),满足fda对亚硝胺杂质的限量要求。
[0130]
实施例7
[0131]
1、伐尼克兰中间体的制备
[0132]
①
、将3kg化合物a(为了更好地保证产品质量,一般可以要求:化合物a中亚硝胺杂质化合物p06的含量≤500ppm,检测值为485ppm)和0.35kg钯炭(pd/c,pd 5%)催化剂,加入到36kg异丙醇和15kg纯化水的反应釜中,控制反应温度在25~35℃,通氢气并搅拌进行反应,hplc检测反应液中化合物a的剩余量≤0.5%停止反应,垫硅藻土过滤除去钯炭催化剂,得到含化合物b的反应液;
[0133]
②
、在惰性气体(氮气)保护下,向步骤
①
所得的反应液中,加入75g碳酸氢钠,然后缓慢加入乙二醛的水溶液(0.56kg乙二醛和6.5kg水)中,控制反应温度在20~30℃,搅拌反应,hplc检测反应液中化合物b的剩余量≤0.5%停止反应,减压蒸馏(真空度≤-0.09mpa,40~50℃)至基本无馏分产生,加入90kg纯化水,控制釜内温度在20~30℃,保温2h,之后离心,干燥(45~50℃),得到化合物c。
[0134]
2、酒石酸伐尼克兰的制备
[0135]
a、在惰性气体(氮气)保护下,向反应瓶中加入20kg纯化水、0.81kg氢氧化钠、8kg二氯甲烷和2kg化合物c(前一步骤制得),控制反应温度在20~35℃,hplc检测反应液中化合物c的剩余量≤0.5%停止反应,之后向反应釜内加入18.5kg二氯甲烷,搅拌,静置,分液,取有机相,水相用二氯甲烷(15kg
×
2)萃取,合并有机相,有机相中加入5kg无水硫酸钠干燥,过滤,对滤液减压蒸馏(真空度≤-0.09mpa,40~50℃)至基本无馏分产生,加入10kg无水乙醇,继续减压蒸馏(真空度≤-0.09mpa,40~50℃)至基本无馏分产生,得到含伐尼克兰的母液;
[0136]
b、向步骤a所得含伐尼克兰的母液中,缓慢加入30kg无水乙醇和1.16kg的l-(+)-酒石酸,控制反应温度在20~30℃,搅拌反应16h,然后停止反应,之后离心,干燥(70~75℃),得到酒石酸伐尼克兰;
[0137]
再按照每克酒石酸伐尼克兰的溶剂用量8ml,将制得的酒石酸伐尼克兰加入到打浆溶剂中,于30~40℃打浆1.5h,之后离心,干燥,即可;经检测,乙酸乙酯打浆处理后的酒石酸伐尼克兰中,亚硝胺杂质化合物p08的含量为3.04ppm,亚硝胺杂质化合物p06和p07的含量为未检出(小于检测限)。
[0138]
实施例8
[0139]
酒石酸伐尼克兰的制备
[0140]
a、在惰性气体(氮气)保护下,向反应瓶中加入20kg纯化水、0.81kg氢氧化钠、8kg二氯甲烷和2kg化合物c(实施例7制得),控制反应温度在20~35℃,hplc检测反应液中化合物c的剩余量≤0.5%停止反应,之后向反应釜内加入18.5kg二氯甲烷,搅拌,静置,分液,取有机相,水相用二氯甲烷(15kg
×
2)萃取,合并有机相,有机相中加入5kg无水硫酸钠干燥,过滤,对滤液减压蒸馏(真空度≤-0.09mpa,40~50℃)至基本无馏分产生,加入10kg无水乙醇,继续减压蒸馏(真空度≤-0.09mpa,40~50℃)至基本无馏分产生,得到含伐尼克兰的母液;
[0141]
b、向步骤a所得含伐尼克兰的母液中,缓慢加入30kg无水乙醇和1.16kg的l-(+)-酒石酸,控制反应温度在20~30℃,搅拌反应16h,然后停止反应,之后离心,干燥(70~75℃),得到酒石酸伐尼克兰;
[0142]
再按照每克酒石酸伐尼克兰的溶剂用量8ml,将制得的酒石酸伐尼克兰加入到打浆溶剂中,于30~40℃打浆1.5h,之后离心,干燥,即可;经检测,二氯甲烷打浆处理后的酒石酸伐尼克兰中,亚硝胺杂质化合物p08的含量为4.47ppm,亚硝胺杂质化合物p06和p07的含量为未检出(小于检测限)。
[0143]
实施例9
[0144]
酒石酸伐尼克兰的制备
[0145]
a、在惰性气体(氮气)保护下,向反应瓶中加入20kg纯化水、0.81kg氢氧化钠、8kg二氯甲烷和2kg化合物c(实施例7制得),控制反应温度在20~35℃,hplc检测反应液中化合物c的剩余量≤0.5%停止反应,之后向反应釜内加入18.5kg二氯甲烷,搅拌,静置,分液,取有机相,水相用二氯甲烷(15kg
×
2)萃取,合并有机相,有机相中加入5kg无水硫酸钠干燥,过滤,对滤液减压蒸馏(真空度≤-0.09mpa,40~50℃)至基本无馏分产生,加入10kg无水乙醇,继续减压蒸馏(真空度≤-0.09mpa,40~50℃)至基本无馏分产生,得到含伐尼克兰的母液;
[0146]
b、向步骤a所得含伐尼克兰的母液中,缓慢加入30kg无水乙醇和1.16kg的l-(+)-酒石酸,控制反应温度在20~30℃,搅拌反应16h,然后停止反应,之后离心,干燥(70~75℃),得到酒石酸伐尼克兰;
[0147]
再按照每克酒石酸伐尼克兰的溶剂用量8ml,将制得的酒石酸伐尼克兰加入到打浆溶剂中,于30~40℃打浆1.5h,之后离心,干燥,即可;经检测,异丙醇打浆处理后的酒石酸伐尼克兰中,亚硝胺杂质化合物p08的含量为2.89ppm,亚硝胺杂质化合物p06和p07的含量为未检出(小于检测限)。
[0148]
实施例10
[0149]
酒石酸伐尼克兰的制备
[0150]
a、在惰性气体(氮气)保护下,向反应瓶中加入20kg纯化水、0.81kg氢氧化钠、8kg二氯甲烷和2kg化合物c(实施例7制得),控制反应温度在20~35℃,hplc检测反应液中化合物c的剩余量≤0.5%停止反应,之后向反应釜内加入18.5kg二氯甲烷,搅拌,静置,分液,取有机相,水相用二氯甲烷(15kg
×
2)萃取,合并有机相,有机相中加入5kg无水硫酸钠干燥,过滤,对滤液减压蒸馏(真空度≤-0.09mpa,40~50℃)至基本无馏分产生,加入10kg无水乙
醇,继续减压蒸馏(真空度≤-0.09mpa,40~50℃)至基本无馏分产生,得到含伐尼克兰的母液;
[0151]
b、向步骤a所得含伐尼克兰的母液中,缓慢加入30kg无水乙醇和1.16kg的l-(+)-酒石酸,控制反应温度在20~30℃,搅拌反应16h,然后停止反应,之后离心,干燥(70~75℃),得到酒石酸伐尼克兰;
[0152]
再按照每克酒石酸伐尼克兰的溶剂用量8ml,将制得的酒石酸伐尼克兰加入到打浆溶剂中,于30~40℃打浆1.5h,之后离心,干燥,即可;经检测,正庚烷打浆处理后的酒石酸伐尼克兰中,亚硝胺杂质化合物p08的含量为24.29ppm。
[0153]
实施例11
[0154]
酒石酸伐尼克兰的制备
[0155]
a、在惰性气体(氮气)保护下,向反应瓶中加入20kg纯化水、0.81kg氢氧化钠、8kg二氯甲烷和2kg化合物c(实施例7制得),控制反应温度在20~35℃,hplc检测反应液中化合物c的剩余量≤0.5%停止反应,之后向反应釜内加入18.5kg二氯甲烷,搅拌,静置,分液,取有机相,水相用二氯甲烷(15kg
×
2)萃取,合并有机相,有机相中加入5kg无水硫酸钠干燥,过滤,对滤液减压蒸馏(真空度≤-0.09mpa,40~50℃)至基本无馏分产生,加入10kg无水乙醇,继续减压蒸馏(真空度≤-0.09mpa,40~50℃)至基本无馏分产生,得到含伐尼克兰的母液;
[0156]
b、向步骤a所得含伐尼克兰的母液中,缓慢加入30kg无水乙醇和1.16kg的l-(+)-酒石酸,控制反应温度在20~30℃,搅拌反应16h,然后停止反应,之后离心,干燥(70~75℃),得到酒石酸伐尼克兰;
[0157]
再按照每克酒石酸伐尼克兰的溶剂用量8ml,将制得的酒石酸伐尼克兰加入到打浆溶剂中,于30~40℃打浆1.5h,之后离心,干燥,即可;经检测,乙醇打浆处理后的酒石酸伐尼克兰中,亚硝胺杂质化合物p08的含量为2.60ppm,亚硝胺杂质化合物p06和p07的含量为未检出(小于检测限)。
[0158]
实施例12-14
[0159]
通过进一步控制本发明起始原料中亚硝胺杂质化合物的含量(例如:将化合物a中亚硝胺杂质化合物p06的含量控制在≤250ppm、≤200ppm、≤150ppm、≤100ppm或更低的范围内),以及增加打浆处理的次数等,我们还制备得到了亚硝胺杂质化合物p08含量分别为0.511ppm、0.205ppm、0.118ppm的酒石酸伐尼克兰。
[0160]
若化合物a中亚硝胺杂质化合物p06的含量过高,可以通过以下打浆处理方法降低p06的含量,例如:将3.3kg化合物a(亚硝胺杂质化合物p06的含量为722ppm)加入到打浆溶剂(8.00kg(11.7l)正庚烷和2.70kg(3l)乙酸乙酯)中,于15~35℃打浆搅拌4~5h,之后离心,水洗滤饼,再离心,干燥,即可。经检测,打浆处理后化合物a中的亚硝胺杂质化合物p06含量为0.03%(315ppm)。这说明,通过一次上述打浆处理能够将化合物a中亚硝胺杂质化合物p06的含量至少降低一半以上。
[0161]
打浆处理的工作原理是:利用打浆溶剂能够溶解目标化合物中的微量杂质使其留在液体中,而不溶的目标化合物仍为固体形式,再通过固液分离,进而降低目标化合物中的杂质含量。
[0162]
实施例15
[0163]
uplc-ms/ms检测酒石酸伐尼克兰原料药中的亚硝胺杂质化合物p08,进行方法学验证。
[0164]
1、溶液配制
[0165]
稀释剂:纯化水
[0166]
对照品储备液:称取19.970mg亚硝胺化合物p08(纯度≥98%),置于10ml量瓶中,加入8ml乙腈,后加入稀释剂溶解并稀释至刻度,混匀。
[0167]
对照品母液:量取对照品储备液37.5μl于10ml量瓶中,稀释剂稀释至刻度,混匀。
[0168]
2、系统适用性
[0169]
系统适用性溶液(100%限度水平对照品溶液):量取对照品母液约100.0μl于10ml量瓶中,稀释剂稀释至刻度,混匀。
[0170]
连续进6针系统适用性溶液,计算6针系统适用性溶液中目标物峰面积的rsd,结果是4%,系统适用性良好。
[0171]
3、线性
[0172]
标准曲线溶液:分别量取对照品母液50.0μl、100.0μl、150.0μl、200.0μl置于4个10ml量瓶中,稀释剂稀释至刻度,混匀,依次作为线性溶液l-3、l-4、l-5、l-6。量取l-3溶液800μ1至10ml量瓶中,稀释剂稀释至刻度,混匀,作为l-2。量取l-2溶液1000μ1至10ml量瓶中,稀释剂稀释至刻度,混匀,作为l-1。
[0173]
取标准曲线溶液分别进样分析,以目标物的浓度(x)为横坐标(单位:ng/ml),峰面积(y)为纵坐标,进行线性回归,计算得到线性方程和线性相关系数r;结果显示,线性方程:y=7370x-12900,线性相关系数r=0.999,线性良好,在0.2951ng/ml~147.6ng/ml(即:0.02951ppm~14.76ppm)的浓度范围内呈良好的线性。
[0174]
4、检测限(lod)
[0175]
lod溶液:量取l-1溶液5000μ1至10ml量瓶中,稀释剂稀释至刻度,混匀,作为lod溶液。
[0176]
取lod溶液连续进样2针,目标峰的s/n(信噪比)分别为11、8,满足不小于3的要求,方法灵敏;lod=0.01476ppm。
[0177]
5、定量限(loq)
[0178]
取定量限溶液(l-1溶液)连续进样6针,目标峰的s/n分别为23、24、17、12、24、14,满足不小于10的要求,方法灵敏;loq=0.02951ppm。
[0179]
6、准确度
[0180]
样品溶液:称取样品(例如:购买或合成的酒石酸伐尼克兰原料药)约100mg置于10ml量瓶中,加稀释剂稀释至刻度,混匀。平行制备2份。
[0181]
准确度溶液:称取样品约100mg置于10ml量瓶中,分别加入对照品母液50.0μl(50%限度水平)、100.0μl(100%限度水平)、150.0μl(150%限度水平),再用稀释剂稀释至刻度,混匀,即得。每个准确度水平平行配制3份。
[0182]
结果显示,在50%限度水平准确度溶液的回收率在95%~102%之间,rsd为4%;在100%限度水平准确度溶液的回收率在90%~103%之间,rsd为7%;在150%限度水平准确度溶液的回收率在98%~100%之间,rsd为1%,符合接受标准,准确度良好。
[0183]
7、重复性
[0184]
称取样品约100mg置于10ml量瓶中,加入对照品母液100.0μl,稀释剂稀释至刻度,混匀。平行配制6份。
[0185]
结果显示,6份重复性溶液中亚硝胺杂质化合物p08含量的rsd为6%,符合接受标准,重复性良好。
[0186]
8、专属性
[0187]
取空白溶液(稀释剂)、100%限度水平对照品溶液、样品溶液(与“重复性”的样品溶液相同)、100%限度水平准确度溶液分别进样分析。
[0188]
结果显示,样品溶液、100%限度水平对照品溶液、100%限度水平准确度溶液的目标物保留时间均为3.47min,目标物的出峰位置处没有明显的干扰,满足分离度的要求,专属性良好。
[0189]
9、中间精密度
[0190]
中间精密度溶液:称取样品约100mg置于10ml量瓶中,加入对照品母液100.0μl,稀释剂稀释至刻度,混匀。平行配制6份。再加上“7、重复性”的6份溶液。
[0191]
计算6份中间精密度溶液和6份重复性溶液共计12份溶液的浓度rsd,为9%,满足不大于15%的要求,符合接受标准,中间精密度良好。
[0192]
10、溶液稳定性
[0193]
取100%限度水平准确度溶液15℃放置,每隔一段时间进样分析,计算每个时间点与零点的浓度比值,21h内该比值在96%~113%之间。
[0194]
11、耐用性
[0195]
分别考察色谱柱温度变化
±
2℃、有机相初始比例
±
1%、流速变化
±
0.01ml/min时,100%限度水平准确度溶液中目标物的浓度值,每个条件下分别进样2针,计算同一参数波动中,6份溶液中目标物浓度值的rsd在4%~6%之间,方法耐用性良好。
[0196]
按照以下公式,计算样品(酒石酸伐尼克兰原料药)中的亚硝胺杂质化合物p08含量,检测2次,取平均值:
[0197]
p08含量:c(ppm)=样品溶液中的p08浓度(ng/ml)/[m(mg)/v(ml)]
[0198]
m:样品质量,mg;
[0199]
v:样品稀释体积,ml。
[0200]
实施例16
[0201]
采用如表11所述条件的液相色谱-串联质谱法,用于本发明酒石酸伐尼克兰原料药中亚硝胺杂质化合物p07的定性和/或定量检测,并进行和通过了方法学验证。
[0202]
表11、检测亚硝胺杂质化合物p07的色谱条件以及质谱条件
[0203][0204]
1、溶液配制
[0205]
空白(稀释剂):水-乙腈(95:5)
[0206]
对照品储备液:称取25mg亚硝胺化合物p07(纯度≥95%),置于50ml容量瓶中,加入乙腈溶解,定容至刻度,混匀(定溶液ⅰ)。取1.0ml定溶液ⅰ,置于100ml容量瓶中,加入稀释剂定容至刻度,混匀,得到对照品储备液。
[0207]
对照品溶液:取0.45ml对照品储备液,置于100ml容量瓶中,加入稀释剂定容至刻度,混匀,即得。
[0208]
样品溶液:取酒石酸伐尼克兰原料药约100mg,置于10ml容量瓶中,加入稀释剂溶解并定容至刻度,混匀。
[0209]
2、系统适用性
[0210]
连续5针对照品溶液,亚硝胺杂质化合物p07峰面积的rsd≤15%,保留时间的rsd≤2.0%,与相邻峰能很好地分开,系统适用性良好。
[0211]
3、线性
[0212]
配制浓度分别为0.002688μg/ml、0.020157μg/ml、0.033594μg/ml、0.053751μg/ml、0.067189μg/ml、0.100783μg/ml(以亚硝胺化合物p07计)的对照品溶液;
[0213]
取上述不同浓度的对照品溶液分别进样分析,以目标物的浓度(x)为横坐标(单位:μg/ml),峰面积(y)为纵坐标,进行线性回归,计算得到线性方程和线性相关系数r;结果显示,线性方程:y=3000000x+2671.7,线性相关系数r=0.998,线性良好,在0.002688μg/
ml~0.100783μg/ml(即:0.2688ppm~10.0783ppm)的浓度范围内呈良好的线性。
[0214]
4、检测限(lod)
[0215]
lod=0.1344ppm,目标峰的s/n(信噪比)为14.8、13.1等,满足大于3的要求,方法灵敏。
[0216]
5、定量限(loq)
[0217]
loq=0.2688ppm,目标峰的s/n为20.5、24.9等,满足大于10的要求,方法灵敏。
[0218]
6、专属性
[0219]
结果显示,亚硝胺杂质化合物p07的保留时间为14.487min左右,相邻峰之间没有干扰,满足分离度的要求,专属性良好。
[0220]
此外,
[0221]
经验证,该检测方法的准确度、精密度、重复性、耐用性等也都符合方法学验证的规定和要求,能够很好地实现酒石酸伐尼克兰原料药中亚硝胺杂质化合物p07的定性和/或定量检测。
[0222]
实施例17
[0223]
采用如表12所述条件的液相色谱-串联质谱法,用于本发明酒石酸伐尼克兰原料药中亚硝胺杂质化合物p06的定性和/或定量检测,并进行和通过了方法学验证。
[0224]
表12、检测亚硝胺杂质化合物p06的色谱条件以及质谱条件
[0225][0226]
1、溶液配制
[0227]
空白(稀释剂):纯化水
[0228]
对照品储备液:称取25mg亚硝胺化合物p06(纯度≥92%),置于50ml容量瓶中,加入乙腈溶解,定容至刻度,混匀(定溶液ⅱ)。取1.0ml定溶液ⅱ,置于100ml容量瓶中,加入稀释剂定容至刻度,混匀,得到对照品储备液。
[0229]
对照品溶液:取4.5ml对照品储备液置于50ml容量瓶中,加入稀释剂定容至刻度,
混匀,即得。
[0230]
样品溶液:取酒石酸伐尼克兰原料药约1000mg,置于20ml顶空瓶中,准确量取5.0ml稀释剂溶解,混匀。
[0231]
2、系统适用性
[0232]
连续5针对照品溶液,亚硝胺杂质化合物p06峰面积的rsd≤15%,保留时间的rsd≤2.0%,与相邻峰能很好地分开,系统适用性良好。
[0233]
3、线性
[0234]
取不同浓度的对照品溶液分别进样分析,以目标物的浓度(x)为横坐标(单位:μg/ml),峰面积(y)为纵坐标,进行线性回归,计算得到线性方程和线性相关系数r;结果显示,线性相关系数r>0.99,线性关系良好。
[0235]
4、检测限(lod)
[0236]
lod=0.0145ppm,目标峰的s/n(信噪比)为5.8,满足大于3的要求,方法灵敏。
[0237]
5、定量限(loq)
[0238]
loq=0.0289ppm,目标峰的s/n为12.0,满足大于10的要求,方法灵敏。
[0239]
6、专属性
[0240]
结果显示,亚硝胺杂质化合物p06的保留时间为14.648min左右,相邻峰之间没有干扰,满足分离度的要求,专属性良好。
[0241]
此外,
[0242]
经验证,该检测方法的准确度、精密度、重复性、耐用性等也都符合方法学验证的规定和要求,能够很好地实现酒石酸伐尼克兰原料药中亚硝胺杂质化合物p06的定性和/或定量检测。
[0243]
实施例18
[0244]
为了更好地保证酒石酸伐尼克兰原料药或制剂中亚硝胺杂质化合物(包括:p06、p07、p08;其中,p08是由p06经中间产物p07转化而来;因此,p08是酒石酸伐尼克兰原料药或制剂中亚硝胺杂质的主要存在形式,p06是化合物a等原料中亚硝胺杂质的主要存在形式,p07是制备过程中的中间产物)的限量要求,需要对起始原料化合物a中的亚硝胺杂质化合物p06进行定性和/或定量检测,例如:采用高效液相色谱法,其色谱条件见表13。
[0245]
表13、检测化合物a中亚硝胺杂质化合物p06的色谱条件
[0246][0247]
1、制作标准曲线
[0248]
空白(稀释剂):乙腈-水(60:40)
[0249]
对照品溶液:称取25mg亚硝胺化合物p06(纯度≥92%),置于25ml容量瓶中,加入稀释剂溶解并定容至刻度,混匀,得到对照品溶液。
[0250]
分别配制浓度为0.0001394mg/ml、0.0004645mg/ml、0.0007432mg/ml、0.000929mg/ml、0.001115mg/ml、0.001394mg/ml、0.001858mg/ml(以亚硝胺化合物p06计)的对照品溶液;
[0251]
取上述不同浓度的对照品溶液分别进样分析,以目标物的浓度(x)为横坐标(单位:mg/ml),峰面积(y)为纵坐标,进行线性回归,计算得到线性方程和线性相关系数r;结果显示,线性方程:y=228.94x+0.0025,线性相关系数r=0.999,线性良好,在该浓度范围内呈良好的线性。
[0252]
检测限:lod=0.004%(即40ppm),目标峰的s/n(信噪比)为6.4,满足大于3的要求,方法灵敏。
[0253]
定量限:loq=0.014%(即140ppm),目标峰的s/n为18.9,满足大于10的要求,方法灵敏。
[0254]
专属性:样品和对照品的混合溶液检测结果显示,亚硝胺杂质化合物p06的保留时间为4.592min左右(分离度r=13.18),化合物a的保留时间为7.587min左右(分离度r=6.82),杂质m01的保留时间为9.373min左右(分离度r=7.24),杂质m02的保留时间为12.45min左右(分离度r=5.25),各个峰之间没有干扰,满足分离度的要求,专属性良好。
[0255]
此外,
[0256]
经验证,该检测方法的准确度、精密度、重复性、耐用性等也都符合方法学验证的规定和要求,能够用于化合物a中亚硝胺杂质化合物p06的定性和/或定量检测。
[0257]
2、对样品进行检测
[0258]
样品溶液:取化合物a约25mg,置于25ml容量瓶中,加入稀释剂溶解并定容至刻度,混匀。
[0259]
样品溶液进样检测,得到其峰面积数据以及色谱图(见:图1),将该峰面积数据代
入上述由对照品溶液得到的线性方程,计算得到检测样品溶液中亚硝胺杂质化合物p06的浓度数据。
[0260]
再按照以下公式,计算样品(化合物a)中的亚硝胺杂质化合物p06含量:
[0261]
p06含量:c(用%或ppm表示)=样品溶液中的p06浓度(mg/ml)/[m(mg)/v(ml)]
[0262]
m:样品质量,mg;
[0263]
v:样品稀释体积,ml。
[0264]
实施例19
[0265]
检测亚硝胺杂质化合物p08是否具有基因毒性(能够致突变或致癌)。
[0266]
1材料和方法
[0267]
1.1实验菌株
[0268]
鼠伤寒沙门氏菌ta97a、ta98、ta100、ta102、ta1535,来源:美国molt ox公司,购自上海宝录生物科技有限公司。采用经鉴定符合要求的鼠伤寒沙门氏菌株进行试验。
[0269]
1.2代谢活化系统
[0270]
大鼠肝s9,来源:上海宝录生物科技有限公司。
[0271]
1.3受试物
[0272]
名称:亚硝胺杂质化合物p08,性状:白色粉末。
[0273]
1.4主要仪器与试剂
[0274]
x sr-204电子天平(瑞士梅特勒公司);高压蒸汽灭菌器(日本sanyo公司);隔水式恒温培养箱(上海一恒科技有限公司);stuart温控摇床培养箱(英国stuart公司);bhc-1300 ii a2生物安全柜(苏净集团苏州安泰空气技术有限公司)。
[0275]
6-磷酸葡萄糖,来源:北京百灵威科技有限公司;辅酶ii,来源:国药集团化学试剂有限公司;营养肉汤,来源:北京陆桥技术股份有限公司;技术琼脂粉,来源:广东环凯微生物科技有限公司;灭菌注射用水,来源:辰欣药业股份有限公司;敌克松,来源:chemservice;甲磺酸甲酯,来源:北京百灵威科技有限公司;2-氨基芴,来源:上海晶纯试剂有限公司;1,8-二羟基蒽醌,来源:美国sigma-aldrich公司;环磷酰胺,来源:美国sigma-aldrich公司;叠氮化钠,来源:riedel-seelie。
[0276]
1.5试验方法(ames试验平板掺入法)
[0277]
实验采用平板掺入法。
[0278]
1.5.l剂量选择与受试物配制
[0279]
剂量设计:按每皿5mg、2.5mg、1.25mg、0.625mg、0.3125mg、0.15625mg进行试验。
[0280]
受试物配制:取本品用二甲基亚砜配成50mg/ml的溶液,用二甲基亚砜依次稀释成25mg/ml、12.5mg/ml、6.25mg/ml、3.125mg/ml、1.5625mg/ml。
[0281]
1.5.2试验方法。
[0282]
将冷冻保存的菌液ta97a、ta98、ta100、ta102、ta1535 0.1ml分别接种于10ml营养肉汤培养基,37℃振荡(100次/min)培养10h。将含0.5mmol/l组氨酸-0.5mmol/l生物素的顶层培养基2.0ml分装于试管中,0.103mpa灭菌20min,50℃水浴保温。然后每管依次加入受试物溶液0.1ml、s9混合液0.5ml(需代谢活化时)和菌液0.1ml,充分混匀,迅速倾入底层琼脂平板上,转动平板,使之分布均匀。水平放置待冷凝固化后,倒置于37℃培养箱里孵育48h,计数每皿回变菌落数。每个剂量均做三个平行板,同时设自发回变组、溶剂对照组和阳性对
照组。
[0283]
1.6试验数据统计
[0284]
数据以均数
±
标准差表示,采用spss 19.0进行数据分析计算。
[0285]
1.7结果判定
[0286]
如果受试物的回变菌落数是溶剂对照回变菌落数的两倍或两倍以上,并呈剂量-反应关系,则该受试物判定为致突变阳性;受试物在任何一个剂量条件下,出现阳性反应并有可重复性,则该受试物判定为致突变阳性。如果受试物经上述试验菌株测定后,无论在加s9和未加s9条件下均为阴性,则该受试物为致突变阴性。
[0287]
2试验结果
[0288]
结果见表14。
[0289]
表14、ames试验结果
[0290][0291]
续表ames试验结果
[0292][0293]
ames试验结果可见,在不加s9的条件下,亚硝胺杂质化合物p08剂量为625μg/皿时,ta1535菌落数超过溶剂对照组的两倍。在加s9条件下,剂量为2500μg/皿时,ta98菌落数超过溶剂对照组的两倍,剂量为5000、2500、1250、625μg/皿时,ta1535菌落数超过溶剂对照组的两倍,并呈剂量-反应关系。这表明,在加与不加代谢活化系统的情况下,亚硝胺杂质化合物p08对鼠伤寒沙门氏菌为致突变阳性。
[0294]
当然,本发明还可以有其它多种实施方式,在不违背本发明精神及其实质的情况下,熟悉本领域的技术人员可以根据本发明作出各种相应的改变和/或变形,这些相应的改变和/或变形都应属于本发明所附的权利要求的保护范围。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1