二炔类化合物及其制备方法与在制备抗肿瘤药物中的应用
二炔类化合物及其制备方法与在制备抗肿瘤药物中的应用
(一)技术领域
1.本发明涉及一种二炔基二硒醚类有机硒化合物及其绿色制备方法和在制备抗肿瘤药物中的应用。
(二)
背景技术:
2.二炔类化合物也是生物、高分子和材料科学中非常重要的中间体和目标产物,是具有生理活性天然产物的重要结构。(a)人参炔醇(b)山参(c)抗hiv 活性。
[0003][0004]
烯二炔类抗肿瘤抗生素分子结构特殊,作用机制新颖,被认为是迄今为止抗肿瘤活性最强的一类抗生素。目前已发现的烯二炔类抗生素有卡奇霉素 (calicheamicin)、新制癌菌素(neocarzinostatin)、力达霉素、达内霉素a (dynemicin a)等。
[0005]
二炔基二硒醚是一类生要的二炔类有机硒化合物,其因含有两个碳碳三键,是较强的富电子基团,因此可以进行亲核加成反应,特别是环加成反应;它们也是引入炔基和硒基制备合成其他有机硒化合物的重要前体,因而在现代有机硒合成中具有重要的地位和应用价值。无论是二硒键还是二炔结构,都被广泛应用于药物的合成,受到医务工作者的青睐。
[0006]
宫颈癌是全球女性中最常见的恶性肿瘤之一。全球范围内,每年有宫颈癌新发病例50万例左右,占所有癌症新发病例的5%。且年轻妇女宫颈癌发病率有明显上升的趋势,因此开发高效低毒的新型化疗药物用于宫颈癌治疗的重要意义。
[0007]
本发明开发出一种二炔基二硒醚化合物绿色制备方法,并将其应用到抑制宫颈癌细胞(hela)当中,开发出具有抗宫颈癌活性化合物。
(三)
技术实现要素:
[0008]
本发明提供一种具有抗肿瘤活性的二炔基二硒醚化合物及其制备方法,本发明提供了一种在相对较低温度下使用较为经济的原料,在乙醇相中合成二炔基二硒醚化合物的方法,该方法具有绿色、经济、高效、无催化剂、反应快、后处理简单等优点。
[0009]
为了实现上述目的,本发明的技术方案如下:
[0010]
第一方面,本发明提供一种式(ⅱ)所示二炔基二硒醚化合物,
[0011][0012]
式(ⅱ)中,r为苯基、被c1~c5烷基、卤素或甲氧基取代的苯基、3
‑
噻吩基、苄基或c4~c8烷基,优选为苯基、被c1~c5烷基、卤素或甲氧基取代的苯基、 3
‑
噻吩基或苄基。
[0013]
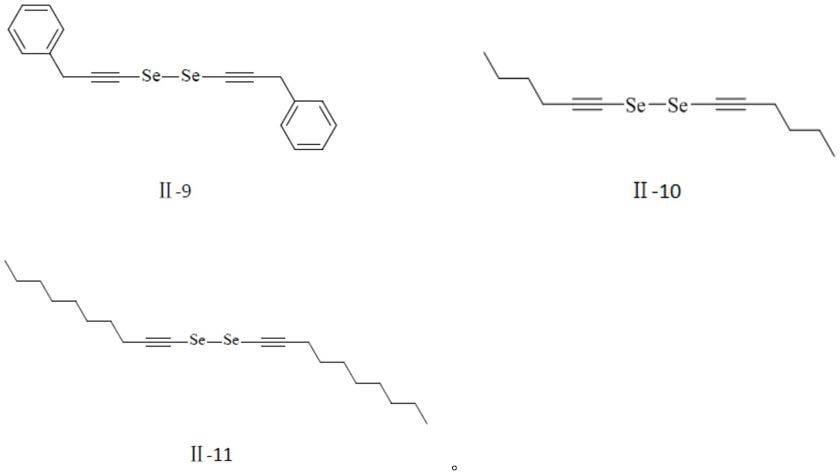
进一步优选地,式(ⅱ)所示二炔基二硒醚化合物为下列化合物之一:
[0014]
[0015][0016]
第二方面,本发明还提供上述式(ⅱ)所示二炔基二硒醚化合物的合成方法,所述的合成方法为:
[0017]
以式(ⅰ)所示化合物和se为反应原料(se既做氧化剂也做还原剂,式(ⅰ) 是还原
剂),以cubr为催化剂,在koh的条件下,以乙醇为溶剂,于55
‑
78℃ (优选78℃)反应2
‑
6h(优选4h),所得反应液经后处理,得到式(ⅱ)所示二炔基二硒醚化合物;所述式(i)所示化合物、se、cubr与koh的物质的量之比为 100:50~300:5~30:50~200(优选为100:100~200:10~20:100~150,更特别优选为100:150:10:100);
[0018][0019]
其中,r为苯基、被c1~c5烷基、卤素或甲氧基取代的苯基、3
‑
噻吩基、苄基或c4~c8烷基。
[0020]
反应式如下:
[0021][0022]
进一步,r为苯基、对甲苯基、对氟苯基、4
‑
戊基苯基、3
‑
溴苯基、4
‑
苯甲醚基、3
‑
噻吩基、2
‑
氯苯基、苄基、丁基、辛基。
[0023]
进一步,所述溶剂的体积以式(i)所示化合物的物质的量计为0.5ml/mmol。
[0024]
进一步,所述后处理为:所述反应液蒸去溶剂后加入水,用乙酸乙酯萃取(2 次),合并有机相,旋蒸去除溶剂,所得粗产品以体积比为20:1的正己烷:乙酸乙酯为洗脱剂进行柱层析,收集含目标产物的洗脱液,蒸去洗脱液,得到所述式(ⅱ)所示二炔基二硒醚化合物。
[0025]
第三方面,本发明还提供上述式(ⅱ)所示二炔基二硒醚化合物在制备抗肿瘤药物中的应用。
[0026]
优选地,所述肿瘤为宫颈癌(hela细胞)。
[0027]
进一步,所述式(ⅱ)所示二炔基二硒醚化合物为下列化合物之一:
[0028][0029]
优选地,所述式(ⅱ)所示二炔基二硒醚化合物为化合物
ⅱ‑
1、
ⅱ‑
2、
ⅱ‑
4、
ⅱ‑
7或
ⅱ‑
8,进一步优选
ⅱ‑
2、
ⅱ‑
4、
ⅱ‑
7或
ⅱ‑
8,特别优选为
ⅱ‑
7。
[0030]
本发明的反应进程可采用常规方法进行监测,例如用tlc或者gc
‑
ms监测原料式(i)所示化合物反应完全来判断反应结束时间点;反应时间通常在3~4h。
[0031]
与现有技术相比,本发明的有益效果在于:
[0032]
(1)本发明合成了一类新颖的具有抗肿瘤活性的二炔基二硒醚类化合物,合成路线短,只需要一步;操作过程简单,产率高。
[0033]
(2)该方法反应不需要无水无氧条件,不需要高温条件,反应在比较温和条件下即可进行,反应时间短,4小时即可完成反应。
[0034]
(3)直接利用硒粉为硒源,与其它硒源相比,硒粉具有廉价、无毒无味的显著优点,绿色环保,更加方便于实验或大生产操作。
[0035]
(4)在有机溶剂当中,乙醇是生物体糖代谢的产物,几乎无毒,被化学家们认为“绿色溶剂”;本发明提供的方法以乙醇作为反应的介质,反应后易于加收再用。
(四)具体实施方式
[0036]
下面结合具体实施例对本发明进行进一步描述,但本发明的保护范围并不仅限于此。
[0037]
本发明中,紫杉醇(cas:33069
‑
62
‑
4)购于萨恩化学技术(上海)有限公司, hela细胞购于武汉普诺赛生命科技有限公司,cck8试剂盒购于武汉菲恩生物科技有限公司。
[0038]
实施例1:二炔基二硒醚类化合物
ⅱ‑
1的制备
[0039]
反应式如下:
[0040][0041]
向反应瓶中加入硒粉119mg(1.5mmol)、氢氧化钾56mg(1mmol)、溴化亚铜14mg(0.1mmol),再加入0.5ml乙醇,然后在搅拌状态下将102mg苯乙炔(1mmol)i
‑
1加入到反应瓶中,在78℃条件下反应4h,停止反应。蒸去乙醇,加入3ml水,反应液用乙酸乙酯(2ml
×
2)萃取2次,合并有机相,旋转蒸发仪蒸去乙酸乙酯,残余物通过柱层析分离提纯,洗脱剂为正己烷:乙酸乙酯=20:1,收集含目标产物的洗脱液,蒸去洗脱液,得到二炔基二硒醚化合物
ⅱ‑
1 166mg,产率92%。gc检测纯度为97.0%。化合物式
ⅱ‑
1的结构表征如下:
[0042]1h nmr(500mhz,cdcl3)δ7.56(dd,j=6.5,1.7hz,4h),7.43
–
7.33(m, 6h);
13
c nmr(126mhz,cdcl3)δ132.50,129.20,128.44,121.80,81.57,73.94.gc
‑ꢀ
ms(ei):m/z 361.9[m+].
[0043]
实施例2:二炔基二硒醚类化合物
ⅱ‑
2的制备
[0044]
反应式如下:
[0045][0046]
向反应瓶中加入硒粉119mg(1.5mmol)、氢氧化钾56mg(1mmol)、溴化亚铜14mg(0.1mmol),再加入0.5ml乙醇,然后在搅拌状态下将116mg对甲基苯乙炔(1mmol)i
‑
2加入到反应瓶中,在78℃条件下反应4h,停止反应。蒸去乙醇,加入3ml水,反应液用乙酸乙酯(2ml
×
2)萃取2次,合并有机相,旋转蒸发仪蒸去乙酸乙酯,残余物通过柱层析分离提纯,洗脱剂
为正己烷:乙酸乙酯=20:1,收集含目标产物的洗脱液,蒸去洗脱液,得到二炔基二硒醚化合物
ⅱꢀ‑
2 178mg,产率92%。gc检测纯度为96.5%。化合物式
ⅱ‑
2的结构表征如下:
[0047]1h nmr(500mhz,cdcl3)δ7.52(d,j=8.2hz,4h),7.17(d,4h),2.37(s, 6h);
13
c nmr(126mhz,cdcl3)δ141.50,136.54,129.24,127.66,115.94,113.07, 21.20.gc
‑
ms(ei):m/z 389.9[m+].
[0048]
实施例3:二炔基二硒醚类化合物
ⅱ‑
3的制备
[0049]
反应式如下:
[0050][0051]
向反应瓶中加入硒粉119mg(1.5mmol)、氢氧化钾56mg(1mmol)、溴化亚铜14mg(0.1mmol),再加入0.5ml乙醇,然后在搅拌状态下将172mg 4
‑
戊基苯乙炔(1mmol)i
‑
3加入到反应瓶中,在78℃条件下反应4h,停止反应。蒸去乙醇,加入3ml水,反应液用乙酸乙酯(2ml
×
2)萃取2次,合并有机相,旋转蒸发仪蒸去乙酸乙酯,残余物通过柱层析分离提纯,洗脱剂为正己烷:乙酸乙酯=20:1,收集含目标产物的洗脱液,蒸去洗脱液,得到二炔基二硒醚化合物
ⅱꢀ‑
3 210mg,产率84%。gc检测纯度为94.7%。化合物式
ⅱ‑
3的结构表征如下:
[0052]1h nmr(500mhz,cdcl3)δ7.55(d,j=8.2hz,4h),7.19(d,j=8.2hz, 4h),2.62(q,j=6.9,6.3hz,4h),1.70
–
1.59(m,4h),1.43
–
1.27(m,8h),0.93(t,j =6.9hz,6h).
13
c nmr(126mhz,cdcl3)δ143.49,128.84,128.55,127.62,116.02, 113.93,35.59,31.45,30.99,22.52,14.01;gc
‑
ms(ei):m/z 502.1[m+].
[0053]
实施例4:二炔基二硒醚类化合物
ⅱ‑
4的制备
[0054]
反应式如下:
[0055][0056]
向反应瓶中加入硒粉119mg(1.5mmol)、氢氧化钾56mg(1mmol)、溴化亚铜14mg(0.1mmol),再加入0.5ml乙醇,然后在搅拌状态下将132mg 4
‑
乙炔基苯甲醚(1mmol)i
‑
4加入到反应瓶中,在78℃条件下反应4h,停止反应。蒸去乙醇,加入3ml水,反应液用乙酸乙酯(2ml
×
2)萃取2次,合并有机相,旋转蒸发仪蒸去乙酸乙酯,残余物通过柱层析分离提纯,洗脱剂为正己烷:乙酸乙酯=20:1,收集含目标产物的洗脱液,蒸去洗脱液,得到二炔基二硒醚化合物
ⅱ‑
4 172mg,产率82%。gc检测纯度为96.2%。化合物式
ⅱ‑
4的结构表征如下:
[0057]1h nmr(500mhz,cdcl3)δ7.57(d,j=9.2hz,4h),6.89(d,j=9.2hz, 4h),3.84(s,6h);
13
c nmr(126mhz,cdcl3)δ159.88,141.47,131.90,129.03, 114.95,113.89,55.37;gc
‑
ms(ei):m/z 421.9[m+].
[0058]
实施例5:二炔基二硒醚类化合物
ⅱ‑
5的制备
[0059]
反应式如下:
[0060][0061]
向反应瓶中加入硒粉119mg(1.5mmol)、氢氧化钾56mg(1mmol)、溴化亚铜14mg(0.1mmol),再加入0.5ml乙醇,然后在搅拌状态下将120mg对氟苯乙炔(1mmol)i
‑
5加入到反应瓶中,在78℃条件下反应4h,停止反应。蒸去乙醇,加入3ml水,反应液用乙酸乙酯(2ml
×
2)萃取2次,合并有机相,旋转蒸发仪蒸去乙酸乙酯,残余物通过柱层析分离提纯,洗脱剂为正己烷:乙酸乙酯=20:1,收集含目标产物的洗脱液,蒸去洗脱液,得到二炔基二硒醚化合物
ⅱ‑
5 166mg,产率84%。gc检测纯度为94.8%。化合物式
ⅱ‑
5的结构表征如下:
[0062]1h nmr(500mhz,cdcl3)δ7.61
‑
7.55(m,4h),7.10
–
7.03(m,4h);
13
c nmr(126mhz,dmso
‑
d6)δ162.60(j=250.7hz),129.87(j=7.6hz),124.75(j= 2.6hz),116.04(j=22.7hz),102.02,80.97;gc
‑
ms(ei):m/z 397.9[m+].
[0063]
实施例6:二炔基二硒醚类化合物
ⅱ‑
6的制备
[0064]
反应式如下:
[0065][0066]
向反应瓶中加入硒粉119mg(1.5mmol)、氢氧化钾56mg(1mmol)、溴化亚铜14mg(0.1mmol),再加入0.5ml乙醇,然后在搅拌状态下将181mg 3
‑
溴苯乙炔(1mmol)i
‑
6加入到反应瓶中,在78℃条件下反应4h,停止反应。蒸去乙醇,加入3ml水,反应液用乙酸乙酯(2ml
×
2)萃取2次,合并有机相,旋转蒸发仪蒸去乙酸乙酯,残余物通过柱层析分离提纯,洗脱剂为正己烷:乙酸乙酯=20:1,收集含目标产物的洗脱液,蒸去洗脱液,得到二炔基二硒醚化合物
ⅱ‑
6 223mg,产率86%。gc检测纯度为95.7%。化合物式
ⅱ‑
6的结构表征如下:
[0067]1h nmr(500mhz,cdcl3)δ7.69(s,2h),7.54
‑
7.52(m,2h),7.48
‑
7.46(m, 2h),7.24
‑
7.21(m,2h);
13
c nmr(126mhz,cdcl3)δ135.14,132.59,131.08, 129.90,123.58,122.26,80.50,74.82.gc
‑
ms(ei):m/z 519.7[m+].
[0068]
实施例7:二炔基二硒醚类化合物
ⅱ‑
7的制备
[0069]
反应式如下:
[0070][0071]
向反应瓶中加入硒粉119mg(1.5mmol)、氢氧化钾56mg(1mmol)、溴化亚铜14mg(0.1mmol),再加入0.5ml乙醇,然后在搅拌状态下将137mg 2
‑
氯苯乙炔(1mmol)i
‑
7加入到反应瓶中,在78℃条件下反应4h,停止反应。蒸去乙醇,加入3ml水,反应液用乙酸乙酯(2ml
×
2)萃取2次,合并有机相,旋转蒸发仪蒸去乙酸乙酯,残余物通过柱层析分离提纯,洗脱剂为正己烷:乙酸乙酯=20:1,收集含目标产物的洗脱液,蒸去洗脱液,得到二炔基二硒醚化合物
ⅱ‑
7 180mg,产率84%。gc检测纯度为96.1%。化合物式
ⅱ‑
7的结构表征如下:
[0072]1h nmr(500mhz,cdcl3)δ7.60
‑
7.58(m,2h),7.45
‑
7.43(m,2h),7.33
‑
7.31 (m,2h),7.26
–
7.24(m,2h);
13
c nmr(126mhz,cdcl3)δ136.99,134.39, 130.29,129.47,126.57,121.84,79.44,78.41.gc
‑
ms(ei):m/z 429.8[m+].
[0073]
实施例8:二炔基二硒醚类化合物
ⅱ‑
8的制备
[0074]
反应式如下:
[0075][0076]
向反应瓶中加入硒粉119mg(1.5mmol)、氢氧化钾56mg(1mmol)、溴化亚铜14mg(0.1mmol),再加入0.5ml乙醇,然后在搅拌状态下将108mg 3
‑
乙炔基噻吩(1mmol)i
‑
8加入到反应瓶中,在78℃条件下反应4h,停止反应。蒸去乙醇,加入3ml水,反应液用乙酸乙酯(2ml
×
2)萃取2次,合并有机相,旋转蒸发仪蒸去乙酸乙酯,残余物通过柱层析分离提纯,洗脱剂为正己烷:乙酸乙酯=20:1,收集含目标产物的洗脱液,蒸去洗脱液,得到二炔基二硒醚化合物
ⅱꢀ‑
8 134mg,产率72%gc检测纯度为94.5%。。化合物式
ⅱ‑
8的结构表征如下:
[0077]1h nmr(500mhz,cdcl3)δ7.61(dd,j=3.0,1.2hz,2h),7.30(dd,j=5.0, 3.0hz,2h),7.19(dd,j=5.0,1.2hz,2h);
13
c nmr(126mhz,cdcl3)δ131.22, 130.16,125.60,120.90,76.58,73.54.gc
‑
ms(ei):m/z 373.8[m+].
[0078]
实施例9:二炔基二硒醚类化合物
ⅱ‑
9的制备
[0079]
反应式如下:
[0080][0081]
向反应瓶中加入硒粉119mg(1.5mmol)、氢氧化钾56mg(1mmol)、溴化亚铜14mg
(0.1mmol),再加入0.5ml乙醇,然后在搅拌状态下将116mg 3
‑
苯基
ꢀ‑1‑
丙炔(1mmol)i
‑
9加入到反应瓶中,在78℃条件下反应4h,停止反应。蒸去乙醇,加入3ml水,反应液用乙酸乙酯(2ml
×
2)萃取2次,合并有机相,旋转蒸发仪蒸去乙酸乙酯,残余物通过柱层析分离提纯,洗脱剂为正己烷:乙酸乙酯=20:1,收集含目标产物的洗脱液,蒸去洗脱液,得到二炔基二硒醚化合物
ⅱ‑
9 167mg,产率86%。gc检测纯度为95.3%。化合物式
ⅱ‑
9的结构表征如下:
[0082]1h nmr(500mhz,cdcl3)δ7.39
‑
7.35(m,8h),7.29(tt,j=5.4,3.7hz,2h), 3.73(s,4h);
13
c nmr(126mhz,cdcl3)δ135.54,128.61,127.93,126.84,75.56, 67.27,25.61.gc
‑
ms(ei):m/z 389.9[m+].
[0083]
实施例10:二炔基二硒醚类化合物
ⅱ‑
10的制备
[0084]
反应式如下:
[0085][0086]
向反应瓶中加入硒粉119mg(1.5mmol)、氢氧化钾56mg(1mmol)、溴化亚铜14mg(0.1mmol),再加入0.5ml乙醇,然后在搅拌状态下将82mg 1
‑
己炔 (1mmol)i
‑
10加入到反应瓶中,在78℃条件下反应4h,停止反应。蒸去乙醇,加入3ml水,反应液用乙酸乙酯(2ml
×
2)萃取2次,合并有机相,旋转蒸发仪蒸去乙酸乙酯,残余物通过柱层析分离提纯,洗脱剂为正己烷:乙酸乙酯=20: 1,收集含目标产物的洗脱液,蒸去洗脱液,得到二炔基二硒醚化合物
ⅱ‑
10 142mg,产率89%。gc检测纯度为95.1%。化合物式
ⅱ‑
10的结构表征如下:
[0087]1h nmr(500mhz,cdcl3)δ2.30
–
2.22(m,4h),1.54
–
1.48(m,4h),1.46
–ꢀ
1.38(m,4h),0.91(t,j=7.3hz,6h);
13
c nmr(126mhz,cdcl3)δ77.38,65.24, 30.37,21.89,18.85,13.48.gc
‑
ms(ei):m/z 322.0[m+].
[0088]
实施例11:苯基二硒醚化合物
ⅱ‑
11的制备
[0089]
反应式如下:
[0090][0091]
向反应瓶中加入硒粉119mg(1.5mmol)、氢氧化钾56mg(1mmol)、溴化亚铜14mg(0.1mmol),再加入0.5ml乙醇,然后在搅拌状态下将138mg 1
‑
癸炔(1mmol)i
‑
11加入到反应瓶中,在78℃条件下反应4h,停止反应。蒸去乙醇,加入3ml水,反应液用乙酸乙酯(2ml
×
2)萃取2次,合并有机相,旋转蒸发仪蒸去乙酸乙酯,残余物通过柱层析分离提纯,洗脱剂为正己烷:乙酸乙酯=20: 1,收集含目标产物的洗脱液,蒸去洗脱液,得到二炔基二硒醚化合物
ⅱ‑
11 190mg,产率88%。gc检测纯度为94.8%。化合物式
ⅱ‑
11的结构表征如下:
[0092]1h nmr(500mhz,cdcl3)δ2.35(t,j=7.5hz,4h),1.66
–
1.61(m,4h), 1.33
–
1.27(m,
20h),0.88(t,j=6.8hz,6h).
13
c nmr(126mhz,cdcl3)δ77.46, 65.31,31.84,29.16,29.09,28.88,28.39,22.66,19.22,14.07.gc
‑
ms(ei):m/z 434.1[m+].
[0093]
实施例12:二炔基二硒醚类化合物
ⅱ‑
1的制备
[0094]
反应式如下:
[0095][0096]
向反应瓶中加入硒粉476mg(6.0mmol)、氢氧化钾224mg(4.0mmol)、溴化亚铜56mg(0.4mmol),再加入2ml乙醇,然后在搅拌状态下将408mg苯乙炔(4mmol)i
‑
1加入到反应瓶中,在78℃条件下反应4h,停止反应。蒸去乙醇,加入12ml水,反应液用乙酸乙酯(8ml
×
2)萃取2次,合并有机相,旋转蒸发仪蒸去乙酸乙酯,残余物通过柱层析分离提纯,洗脱剂为正己烷:乙酸乙酯=20:1,收集含目标产物的洗脱液,蒸去洗脱液,得到二炔基二硒醚化合物
ⅱ‑
1 648mg,产率90%。
[0097]
实施例13:二炔基二硒醚类化合物
ⅱ‑
1的制备
[0098]
反应式如下:
[0099][0100]
向反应瓶中加入硒粉119mg(1.5mmol)、氢氧化钾56mg(1mmol)、铜6.4mg (0.1mmol),再加入0.5ml乙醇,然后在搅拌状态下将102mg苯乙炔(1mmol) i
‑
1加入到反应瓶中,在78℃条件下反应4h,停止反应。蒸去乙醇,加入3ml 水,反应液用乙酸乙酯(2ml
×
2)萃取2次,合并有机相,旋转蒸发仪蒸去乙酸乙酯,残余物通过柱层析分离提纯,洗脱剂为正己烷:乙酸乙酯=20:1,收集含目标产物的洗脱液,蒸去洗脱液,得到二炔基二硒醚化合物
ⅱ‑
1 43mg,产率 24%。
[0101]
实施例14:二炔基二硒醚类化合物
ⅱ‑
1的制备
[0102]
反应式如下:
[0103][0104]
向反应瓶中加入硒粉119mg(1.5mmol)、氢氧化钾56mg(1mmol)、氯化铜13.4mg(0.1mmol),再加入0.5ml乙醇,然后在搅拌状态下将102mg苯乙炔(1mmol)i
‑
1加入到反应瓶中,在78℃条件下反应4h,停止反应。蒸去乙醇,加入3ml水,反应液用乙酸乙酯(2ml
×
2)萃取2次,合并有机相,旋转蒸发仪蒸去乙酸乙酯,残余物通过柱层析分离提纯,洗脱剂为正己烷:乙酸乙酯=20: 1,收集含目标产物的洗脱液,蒸去洗脱液,得到二炔基二硒醚化合物
ⅱ‑
1 40mg,产率22%。
[0105]
实施例15:二炔基二硒醚类化合物
ⅱ‑
1的制备
[0106]
反应式如下:
[0107][0108]
向反应瓶中加入硒粉119mg(1.5mmol)、氢氧化钾56mg(1mmol)、溴化亚铜7mg(0.05mmol),再加入0.5ml乙醇,然后在搅拌状态下将102mg苯乙炔(1mmol)i
‑
1加入到反应瓶中,在78℃条件下反应4h,停止反应。蒸去乙醇,加入3ml水,反应液用乙酸乙酯(2ml
×
2)萃取2次,合并有机相,旋转蒸发仪蒸去乙酸乙酯,残余物通过柱层析分离提纯,洗脱剂为正己烷:乙酸乙酯=20: 1,收集含目标产物的洗脱液,蒸去洗脱液,得到二炔基二硒醚化合物
ⅱ‑
1 67mg,产率37%
[0109]
实施例16:二炔基二硒醚类化合物
ⅱ‑
1的制备
[0110]
反应式如下:
[0111][0112]
向反应瓶中加入硒粉119mg(1.5mmol)、氢氧化钾56mg(1mmol)、溴化亚铜28mg(0.2mmol),再加入0.5ml乙醇,然后在搅拌状态下将102mg苯乙炔(1mmol)i
‑
1加入到反应瓶中,在78℃条件下反应3h,停止反应。蒸去乙醇,加入3ml水,反应液用乙酸乙酯(2ml
×
2)萃取2次,合并有机相,旋转蒸发仪蒸去乙酸乙酯,残余物通过柱层析分离提纯,洗脱剂为正己烷:乙酸乙酯=20: 1,收集含目标产物的洗脱液,蒸去洗脱液,得到二炔基二硒醚化合物
ⅱ‑
1 164mg,产率91%。gc检测纯度为95.9%。
[0113]
实施例17:二炔基二硒醚类化合物
ⅱ‑
1的制备
[0114]
反应式如下:
[0115][0116]
向反应瓶中加入硒粉119mg(1.5mmol)、氢氧化钾28mg(0.5mmol)、溴化亚铜14mg(0.1mmol),再加入0.5ml乙醇,然后在搅拌状态下将102mg苯乙炔(1mmol)i
‑
1加入到反应瓶中,在78℃条件下反应6h,停止反应。蒸去乙醇,加入3ml水,反应液用乙酸乙酯(2ml
×
2)萃取2次,合并有机相,旋转蒸发仪蒸去乙酸乙酯,残余物通过柱层析分离提纯,洗脱剂为正己烷:乙酸乙酯=20: 1,收集含目标产物的洗脱液,蒸去洗脱液,得到二炔基二硒醚化合物
ⅱ‑
1 81mg,产率45%
[0117]
实施例18:二炔基二硒醚类化合物
ⅱ‑
1的制备
[0118]
反应式如下:
[0119][0120]
向反应瓶中加入硒粉119mg(1.5mmol)、氢氧化钾112mg(2mmol)、溴化亚铜14mg(0.1mmol),再加入0.5ml乙醇,然后在搅拌状态下将102mg苯乙炔(1mmol)i
‑
1加入到反应瓶中,在78℃条件下反应4h,停止反应。蒸去乙醇,加入3ml水,反应液用乙酸乙酯(2ml
×
2)萃取2次,合并有机相,旋转蒸发仪蒸去乙酸乙酯,残余物通过柱层析分离提纯,洗脱剂为正己烷:乙酸乙酯=20: 1,收集含目标产物的洗脱液,蒸去洗脱液,得到二炔基二硒醚化合物
ⅱ‑
1 162mg,产率90%。gc检测纯度为96.2%。
[0121]
实施例19:二炔基二硒醚类化合物
ⅱ‑
1的制备
[0122]
反应式如下:
[0123][0124]
向反应瓶中加入硒粉119mg(1.5mmol)、碳酸钠106mg(1mmol)、溴化亚铜14mg(0.1mmol),再加入0.5ml乙醇,然后在搅拌状态下将102mg苯乙炔 (1mmol)i
‑
1加入到反应瓶中,在78℃条件下反应4h。利用gc
‑
ms方法检测反应液,没有发现目标产物
ⅱ‑
1生成。
[0125]
实施例20:二炔基二硒醚类化合物
ⅱ‑
1的制备
[0126]
反应式如下:
[0127][0128]
向反应瓶中加入硒粉119mg(1.5mmol)、三乙胺101mg(1mmol)、溴化亚铜14mg(0.1mmol),再加入0.5ml乙醇,然后在搅拌状态下将102mg苯乙炔 (1mmol)i
‑
1加入到反应瓶中,在78℃条件下反应4h,停止反应。利用gc
‑
ms 检测反应液,没有发现目标产物
ⅱ‑
1生成。
[0129]
实施例21:二炔基二硒醚化合物ii对hela细胞抑制活性的测定
[0130]
采用cck8试剂盒检测待测样品对hela细胞的细胞毒性作用来评价二炔基二硒醚化合物ii的抗肿瘤作用。
[0131]
(1)将hela细胞放置在t25细胞培养皿贴壁培养,在10%fbs的dmem 溶液中,5%的co2和37℃条件下培养。待其生长至对数期时,从培养箱中取出 t25细胞培养皿,加入2ml的胰蛋白酶消化。
[0132]
(2)消化完全后,用1ml的移液枪吸出胰蛋白酶,加入1.5ml的ep管中,在1500rpm条件下离心5分钟,弃掉胰蛋白酶,加入培养液悬浮并稀释至培养液的细胞密度为1
×
105/ml。用1ml的移液枪将细胞悬浮液加到96孔板中,每孔的体积为100μl,空白对照加入100μl的不含细胞的培养液,将96孔板放到细胞培养箱中培养过夜。
[0133]
(3)待96孔板的细胞长到细胞对数期时,分别吸取1μl不同浓度(40μg/ml、 20μg/ml、10μg/ml、5μg/ml)的待测样品(ii
‑
1~ii
‑
11和紫杉醇)1μl于实验组中,每个浓度设置三个复孔孵育24h。孵育完后每个孔加入10μl的cck8试剂,在37℃、5%的co2的培养箱中再培养2h。对照组加入1μl含相应浓度的 dmso。培养2h后完后用酶标仪在450nm测量吸光值。用以下公式计算细胞抑制率:
[0134][0135]
二炔基二硒醚化合物ii
‑
1~ii
‑
11和紫杉醇对hela细胞的抑制率列于表1:
[0136]
由表1可以看出:当给药浓度为40μg/ml时,化合物
ⅱ‑
1、
ⅱ‑
2、
ⅱ‑
4、
ⅱꢀ‑
7、
ⅱ‑
8和紫杉醇的抑制率都达到50%以上,具有较好的抑制活性;当给药浓度为20μg/ml时,化合物
ⅱ‑
2、
ⅱ‑
4、
ⅱ‑
7、
ⅱ‑
8和紫杉醇的抑制率都达到50%以上,具有较好的抑制活性;当给药浓度为10μg/ml时,只有化合物
ⅱ‑
7的抑制率达到50%以上,此时紫杉醇的抑制率为33.16%,化合物
ⅱ‑
2和
ⅱ‑
4的抑制率都大于紫杉醇,分别为44.75%和46.05%,具有强的抑制活性;当给药浓度降为 5μg/ml时,紫杉醇的抑制率为13.89%,化合物
ⅱ‑
2、
ⅱ‑
4、
ⅱ‑
7的抑制率都大于紫杉醇,分别为15.94%、20.48%、29.09%,具有强的抑制活性。因此,化合物
ⅱ‑
2、
ⅱ‑
4和
ⅱ‑
7有比紫杉醇强的对hela细胞的抑制活性。
[0137]
表1二炔基二硒醚化合物ii对hela细胞的抑制活性
[0138]
[0139][0140]
“‑‑”
表示抑制率低于10%。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1