一种分离提取鬼针聚炔苷和鬼针聚炔苷B的制备工艺
一种分离提取鬼针聚炔苷和鬼针聚炔苷b的制备工艺
技术领域
1.本发明属于炔苷类化合物提取技术领域,具体涉及一种分离提取鬼针聚炔苷和鬼针聚炔苷b的制备工艺。
背景技术:
2.鬼针草,又名粘身草、婆婆针,属于菊科,鬼针草属,在南美洲、热带及亚热带地区均有分布。民间药用历史悠久,具有清热解毒、散瘀消肿等功效,治疗作用广泛,疗效确切。
3.现代研究表明:鬼针草主含黄酮类、聚炔(苷)类、萜类、酚酸类、奥卡宁类等化合物,而其药理活性则集中体现在诸如抗疟、抗炎、降血脂、降血压、降血糖、抗氧化、抗肿瘤等方面,尤以其对心血管的保护作用及抗氧化作用深受科研人员关注和民间大众认可,现在我国已开发出多种含鬼针草属植物的临床方剂,因此对鬼针草的研究具有潜在的社会价值及科研价值。
4.聚炔类成分是鬼针草含有的一种主要有效成分,在抗微生物、细胞毒及抗肿瘤活性等方面具有良好的生物活性。
5.公开号为cn106939028a的专利文献涉及炔苷类化合物及其提取分离方法和应用,属于天然化合物提取分离领域,该提取分离方法为将桔梗的粉末用甲醇超声提取,将提取液浓缩得第一浸膏;第一浸膏加水溶解并加入到吸附树脂用乙醇进行梯度洗脱,将梯度洗脱的洗脱液第二次浓缩得第二浸膏;将第二浸膏用体积比为1:28
‑
32:1:28
‑
32的正己烷、乙酸乙酯、甲醇和水的溶剂系统进行逆流色谱分离纯化,收集聚炔类成分流出液;将聚炔类成分流出液第三次浓缩后用高效液相色谱仪,以乙腈
‑
水作流动相进行制备;该方法高效、快速,提纯出的炔苷类化合物具有抗微生物、抗肿瘤等活性,能够被应用于制备抗生物毒性药物。
6.公开号为cn106674299a的专利文献涉及一种聚炔苷类化合物及其制备方法和应用,先取兴安独活的干燥根进行回流提取和萃取,得到正丁醇萃取层;将正丁醇萃取层上样于硅胶柱,以氯仿
‑
甲醇系统作为洗脱液,进行梯度洗脱,对流出液进行检测,将洗脱液体积比为5:1的流份合并,除去溶剂,得到第一次过柱部分;将第一次过柱部分上样于反相硅胶层析柱,以甲醇
‑
水系统作为洗脱液,进行梯度洗脱,将洗脱液体积比为45:55的流份合并,除去溶剂,得到第二次过柱部分;将第二次过柱部分上样于高效液相色谱分离柱,用流动相进行等度洗脱,得到聚炔苷类化合物。本发明能够从兴安独活中提取出聚炔苷类化合物,该化合物能够促进细胞对葡萄糖的摄入和转化。
7.上述制备聚炔苷类化合物的方法不能很好地适用于鬼针草中聚炔苷类化合物的提取,同时现有基于鬼针草提取鬼针聚炔苷和鬼针聚炔苷b的收率非常低,亟需改进。
技术实现要素:
8.本发明所要解决的技术问题是,针对现有技术的不足,提供一种分离提取鬼针聚炔苷和鬼针聚炔苷b的制备工艺,以高效提取出鬼针草中含有的鬼针聚炔苷和鬼针聚炔苷
b。
9.为解决上述技术问题,本发明所采用的技术方案是:分离提取鬼针聚炔苷和鬼针聚炔苷b的制备工艺,包括如下步骤:步骤s1:将鬼针草的粉末,采用乙醇浸泡进行渗漉,将渗漉液浓缩得流浸膏,将所述流浸膏经溶剂萃取后,减压回收溶剂,得萃取浸膏;步骤s2:将所述萃取浸膏配制成乙醇混合溶液,离心,取上清液分次上样于大孔吸附树脂柱,采用乙醇进行梯度洗脱,将所述梯度洗脱后的第一洗脱液减压回收溶剂得到洗脱浸膏;步骤s3:将所述洗脱浸膏配制成乙醇混合溶液,离心,取上清液加入到聚酰胺柱,先以适量水冲洗,再以乙醇进行梯度洗脱,得到第二洗脱液;步骤s4:将所述第二洗脱液配制成甲醇混合溶液,分次上样于小孔吸附树脂柱,采用甲醇进行梯度洗脱,得到第三洗脱液;将所述第三洗脱液以葡聚糖凝胶sephadex lh
‑
20进行分离,以甲醇进行洗脱,得到第四洗脱液;步骤s5:将所述第四洗脱液浓缩,以甲醇
‑
水作为流动相,采用制备高效液相系统进行制备。
10.进一步的,所述采用乙醇浸泡进行渗漉,具体为:将鬼针草的粉末置于渗漉桶内,缓缓加入体积分数为75
‑
90%乙醇(例如:75%、80%、85%或90%),直至粉末被全部浸湿且乙醇液面没过药材粉末9
‑
11cm即停止加入,以塑料膜封紧渗漉桶口,浸泡24
‑
72h(例如:24h、36h、48h、64h或72h);之后开始渗漉,以5
‑
10ml/(kg
•
min) [例如:5、6、7、8、9或10 ml/(kg
•
min)]的速度收集渗漉液,抽滤,并随时补足体积分数为75
‑
90%乙醇,得到总的渗漉液。进一步优选地,总的渗漉液的体积量以l计为鬼针草重量以kg计的10~15倍。
[0011]
对于本发明,步骤s1中所述的溶剂为石油醚、乙酸乙酯和正丁醇。
[0012]
进一步的,所述步骤s1中浓缩是在40
‑
47℃温度下旋转蒸发浓缩至无乙醇,得流浸膏。其中,温度可以为40℃、43℃、45℃、46℃、47℃。进一步优选地,流浸膏的体积量与鬼针草的重量之比为1l:(1
‑
1.5)kg。
[0013]
进一步的,所述大孔吸附树脂柱为大孔吸附树脂柱d101、ab
‑
8、hp
‑
20或xad
‑
4。
[0014]
在本发明中,步骤s2中将所述萃取浸膏配制成乙醇混合溶液,是将萃取浸膏与体积分数为15
‑
25%乙醇溶剂,充分混匀。其中,乙醇溶剂的体积分数可选15%、20%、25%。
[0015]
进一步的,所述步骤s2采用乙醇进行梯度洗脱中乙醇的体积浓度为20
‑
100%,例如:20%、40%、60%、80%、100%。
[0016]
对于本发明,步骤s3中将所述洗脱浸膏配制成乙醇混合溶液,是将洗脱浸膏与体积分数为8
‑
12%的乙醇溶剂,充分混匀。其中,乙醇溶剂的体积分数可选8%、10%、12%。
[0017]
对于本发明,步骤s3中所述先以适量水冲洗,其中水的用量可根据需要进行常规确定,适量水可优选为1
‑
10倍柱体积,例如:1倍柱体积的水、5倍柱体积的水或10倍柱体积的水。
[0018]
进一步的,所述步骤s3以乙醇进行梯度洗脱中乙醇的体积浓度为5
‑
95%,例如:5%、10%、20%、30%、40%、50%、60%、75%、80%、95%。
[0019]
对于本发明,步骤s4中将所述第二洗脱液配制成甲醇混合溶液,是将所述第二洗脱液与体积分数为30
‑
40%甲醇溶液,充分混匀,其中甲醇溶液的体积分数可以为30%,35%,
40%。
[0020]
进一步的,所述步骤s4采用甲醇进行梯度洗脱中甲醇的体积浓度为35
‑
65%(例如:35%、40%、45%、50%、55%、60%、65%),所述以甲醇进行洗脱中甲醇的体积浓度为75
‑
85%(例如:75%、80%、85%)。
[0021]
进一步的,所述以甲醇
‑
水作为流动相中甲醇和水的体积比为33:67。
[0022]
鬼针草始载于《本草拾遗》,其味苦,平,无毒。具有清热解毒、散瘀消肿之功效。主治咽喉肿痛、腹泻、疟疾、痢疾、肝炎、急性肾炎、胃痛、噎膈、肠痈、跌打损伤及蛇虫咬伤等病症。鬼针草中化学成分复杂,主要的生物活性成分为黄酮类、聚炔类等。
[0023]
据报道已从鬼针草中分离出鬼针聚炔苷和鬼针聚炔苷b(马明.婆婆针有效部位化学成分的研究.2010.),然而鬼针聚炔苷和鬼针聚炔苷b的收率却极低,还不到0.01
‰
。如此低的收率是其产业化应用的一大屏障,如果无法解决这一问题,无论其药用价值多高,都无法实现更广泛且有效的临床应用,只能局限于原料药的应用层面。因此,提高鬼针聚炔苷和鬼针聚炔苷b的收率是关键。
[0024]
与现有技术相比,本发明的有益效果如下:1)本发明基于对鬼针草特性的研究,采用了渗漉法提取鬼针草中的有效成分,并获得了针对鬼针草的渗漉法提取工艺及其参数,结果表明,鬼针草采用渗漉法提取较之乙醇回流提取,提取效果更佳,目标成分鬼针聚炔苷和鬼针聚炔苷b的收率得以显著提高。
[0025]
2)本发明对渗漉法提取的鬼针草浸膏采用三步不同的洗脱程序,保证更多的杂质被除去,提高了提取物中目标成分的含量。
[0026]
3)本发明目标成分鬼针聚炔苷和鬼针聚炔苷b的收率高,鬼针聚炔苷b的收率高于0.230
‰
,鬼针聚炔苷的收率高于0.150
‰
,较之现有工艺的平均收率提高15倍以上。其中的收率指目标成分的重量与药材重量的百分比。
[0027]
4)本发明目标成分鬼针聚炔苷和鬼针聚炔苷b的纯度高,可达95%以上。
[0028]
5)本发明所用溶剂包括乙酸乙酯、乙醇和甲醇,而常规工艺多采用氯仿
‑
甲醇,氯仿
‑
甲醇
‑
水等的混合体系,回收步骤繁琐,安全性低,故本发明在溶剂的使用上更为安全、环保,效果显著。
[0029]
6)已有研究表明,炔苷类化合物具有抗微生物、细胞毒以及抗肿瘤等良好生物活性,并能够被应用于制备抗生物毒性药物;本发明提取分离方法能够高效、环保、安全地提纯出鬼针草中的炔苷类化合物鬼针聚炔苷和鬼针聚炔苷b,能够进一步满足市场对炔苷类化合物的需求。
[0030]
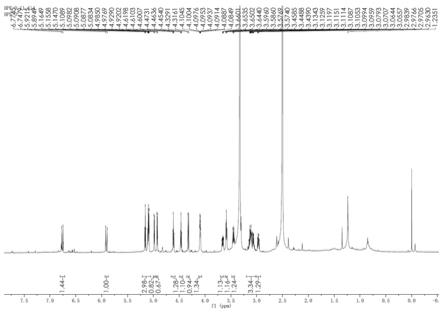
说明书附图图1为本发明化合物1的 1 h
‑
nmr图谱;图2为本发明化合物1的 13 c
‑
nmr图谱;图3为本发明化合物1的 hmbc图谱;图4为本发明化合物2的 13 c
‑
nmr图谱。
具体实施方式
[0031]
为了更好地理解本发明,下面结合实施例进一步清楚阐述本发明的内容,但本发明的保护内容不仅仅局限于下面的实施例。在下文的描述中,给出了大量具体的细节以便
提供对本发明更为彻底的理解。然而,对于本领域技术人员来说显而易见的是,本发明可以无需一个或多个这些细节而得以实施。
[0032]
实施例1取鬼针草,用粉碎机反复粉碎至全部过2号筛(药典粗粉),最后称重共计13.2kg,置于渗漉桶内,缓缓加入体积分数80%的乙醇,直至粉末被全部浸湿且乙醇液面没过药材粉末约10cm即停止加入,以塑料膜封紧渗漉桶口,浸泡48h;之后开始渗漉,以10ml/(kg
•
min)的速度收集渗漉液,抽滤,并随时补足80%乙醇,得到总的渗漉液,总的渗漉液的体积量以l计为鬼针草重量以kg计的10倍;取渗漉液,45℃旋转蒸发仪浓缩至无乙醇,得流浸膏,流浸膏的体积量与鬼针草的重量之比为1l:1kg,经石油醚、乙酸乙酯、正丁醇分别萃取,萃取至颜色变浅后更换溶剂萃取,萃取完成后,减压回收溶剂,得鬼针草各萃取部位,取乙酸乙酯部位浸膏为萃取浸膏;取萃取浸膏(203.68g),配制成20%乙醇溶液,溶液于离心机内4000r/min离心5分钟,取上清液湿法分次上样于5kg d101大孔吸附树脂柱,每次上样量为500ml,上样后,待样品溶液全部进入柱材料,分别以20%
‑
40%
‑
60%
‑
80%
‑
100%乙醇进行梯度洗脱,每个梯度均洗脱至流出液无颜色之后更换下一个梯度;相同的洗脱剂洗脱下来的流分合并,减压回收溶剂后得到相应的浸膏,更换梯度后的流分合并进入下一个梯度,共收集到5个流分段;取40%乙醇流分作为洗脱浸膏;取洗脱浸膏(87.5g),将其配制成10%乙醇溶液,溶液于离心机内4000r/min离心5分钟,取上清液分次湿法上样于3kg聚酰胺柱,每次上样量200ml,上样后,待样品溶液全部流入聚酰胺,以1倍柱体积的水进行冲洗之后,再分别以10%乙醇
‑
50%乙醇
‑
95%乙醇进行洗脱,同样每个梯度均洗脱至流出液基本无颜色后更换下一个梯度;相同的洗脱剂洗脱下来的流分合并,收集共得到3个流分段;取10%乙醇流分作为第二洗脱液;取第二洗脱液,将其配制成35%甲醇溶液,同上述方法一样,将约30g的样品分次上样于小孔吸附树脂(mci)柱1kg,每次上样量50ml;之后分别以35%甲醇
‑
50%甲醇
‑
65%甲醇进行梯度洗脱,分别得到这三个流分段;取50%甲醇流分作为第三洗脱液;继续把第三洗脱液以葡聚糖凝胶sephadexlh20进行分离,以80%甲醇进行洗脱,以自动流分收集器进行收集,每管5ml,收集好后根据聚酰胺薄层色谱板的展开结果进行合并,分别得到a、b、c、d、e5个流分段;取c部位作为第四洗脱液;取第四洗脱液,于45℃旋蒸避光适当浓缩后,通过制备高效液相系统,制备分离出鬼针草中活性单体成分,所需色谱条件为甲醇:水=33:67,而后所得样品于分析液相210nm处初步检识化合物的纯度,待其纯度达到95%以上后,低温避光旋蒸蒸干溶剂,得干燥粉末。
[0033]
通过核磁共振碳谱和氢谱(nmr)、h的异核多碳相关谱(hmbc)以及查阅既往研究文献等分析方法和手段,对分离所得的化合物进行结构鉴定,结果2个化合物1、2分别鉴定为鬼针聚炔苷b(bipinnata polyacetyloside b)、鬼针聚炔苷(bipinnata polyacetyloside),结构式分别为:
化合物1(3135.8mg,收率0.238
‰
)和化合物2(2075.6mg,收率0.157
‰
)均为淡黄色粉末,常温下给予光照放置后显紫色,结合本属已经报道的化学成分,推测其可能为聚炔类成分。
[0034]
化合物1的1h
‑
nmr(600mhz)谱给出以下信息,在低场区可见一个反式双键,δ
h 6.76(1h,dt,j = 16.0,2.3 hz),δ
h 5.91(1h,j = 16.0,2.3 hz),1个葡萄糖端基质子信号δ
h 4.32(1h,d,j = 7.8 hz)。
13
c nmr给出19个碳信号,δ
c 154.6,δ
c 104.9为双键上的两个碳信号;δ
c 102.9为葡萄糖端基碳信号。hmbc谱显示,δ
h 4.61(h
‑
1)与 δ
c 80.2(c
‑
3),δ
c 70.2(c
‑
2)相关;δ
h 6.76(h
‑
12)与δ
c 61.4(c
‑
13)δ
c 104.9(c
‑
11);δ
h 4.32(glu
‑
h
‑
1)与δ
c 70.3(c
‑
2)相关。
[0035]
参阅图1
‑
3,结合氢谱和碳谱数据,与文献对照(马明. 婆婆针有效部位化学成分的研究[d].山东中医药大学,2003.),最终确定化合物1的结构确定为:鬼针聚炔苷b(bipinnata polyacetyloside b),
13
c nmr谱信号归属如表1。
[0036]
参阅图4,化合物2与化合物1类似,
13
c nmr(150mhz)同样给出19个碳信号,与化合物1最大的不同在于δc 150.12(c
‑
3)和δc 109.37(c
‑
4),结合化合物1的信息以及已有研究(马明. 婆婆针有效部位化学成分的研究[d].山东中医药大学,2003.),确定化合物2为鬼针聚炔苷(bipinnata polyacetyloside),其c谱归属如表1所示:表1化合物1、2的c谱归属
实施例2取鬼针草,用粉碎机反复粉碎至全部过2号筛(药典粗粉),最后称重共计10kg,置于渗漉桶内,缓缓加入体积分数90%的乙醇,直至粉末被全部浸湿且乙醇液面没过药材粉末10cm即停止加入,以塑料膜封紧渗漉桶口,浸泡24h;之后开始渗漉,以5ml/(kg
•
min)的速度收集渗漉液,抽滤,并随时补足90%乙醇,得到总的渗漉液,总的渗漉液的体积量以l计为鬼针草重量以kg计的12倍;取渗漉液,40℃旋转蒸发仪浓缩至无乙醇,得流浸膏,流浸膏的体积量与鬼针草的重量之比为1l:1.2kg,经石油醚、乙酸乙酯、正丁醇分别萃取,萃取至颜色变浅后更换溶剂萃取,萃取完成后,减压回收溶剂,得鬼针草各萃取部位,取乙酸乙酯部位浸膏为萃取浸膏;取萃取浸膏(157.85g),配制成15%乙醇溶液,溶液于离心机内4000r/min离心5分钟,取上清液湿法分次上样于4kg ab
‑
8大孔吸附树脂柱,每次上样量为500ml,上样后,待样品溶液全部进入柱材料,分别以20%
‑
40%
‑
60%
‑
80%
‑
100%乙醇进行梯度洗脱,每个梯度均洗脱至流出液无颜色之后更换下一个梯度;相同的洗脱剂洗脱下来的流分合并,减压回收溶剂后得到相应的浸膏,更换梯度后的流分合并进入下一个梯度,共收集到5个流分段;取40%乙醇流分作为洗脱浸膏;取洗脱浸膏(67.9g),将其配制成8%乙醇溶液,溶液于离心机内4000r/min离心5分钟,取上清液分次湿法上样于2.5kg聚酰胺柱,每次上样量200ml,上样后,待样品溶液全部流入聚酰胺,以1倍柱体积的水进行冲洗之后,再分别以5%乙醇
‑
50%乙醇
‑
95%乙醇进行洗脱,同样每个梯度均洗脱至流出液基本无颜色后更换下一个梯度;相同的洗脱剂洗脱下来的流分合并,收集共得到3个流分段;取10%乙醇流分作为第二洗脱液;取第二洗脱液,将其配制成30%甲醇溶液,同上述方法一样,将约23g的样品分次上样于小孔吸附树脂(mci)柱1kg,每次上样量50ml;之后分别以35%甲醇
‑
50%甲醇
‑
65%甲醇进
行梯度洗脱,分别得到这三个流分段;取50%甲醇流分作为第三洗脱液;继续把第三洗脱液以葡聚糖凝胶sephadexlh20进行分离,以75%甲醇进行洗脱,以自动流分收集器进行收集,每管5ml,收集好后根据聚酰胺薄层色谱板的展开结果进行合并,分别得到a、b、c、d、e5个流分段;取c部位作为第四洗脱液;取第四洗脱液,于43℃旋蒸避光适当浓缩后,通过制备高效液相系统,制备分离出鬼针草中活性单体成分,所需色谱条件为甲醇:水=33:67,而后所得样品于分析液相210nm处初步检识化合物的纯度,待其纯度达到95%以上后,低温避光旋蒸蒸干溶剂,得干燥粉末。
[0037]
通过质谱(ms)、核磁共振碳谱和氢谱(nmr)、高效液相色谱(hplc)以及查阅既往研究文献等分析方法和手段,对分离所得的化合物进行结构鉴定,结果2个化合物1、2分别鉴定为鬼针聚炔苷b(bipinnata polyacetyloside b)、鬼针聚炔苷(bipinnata polyacetyloside)。
[0038]
鬼针聚炔苷b的收率为0.243
‰
,鬼针聚炔苷收率为0.161
‰
。
[0039]
实施例3取鬼针草,用粉碎机反复粉碎至全部过2号筛(药典粗粉),最后称重共计15kg,置于渗漉桶内,缓缓加入体积分数75%的乙醇,直至粉末被全部浸湿且乙醇液面没过药材粉末约10cm即停止加入,以塑料膜封紧渗漉桶口,浸泡72h;之后开始渗漉,以8ml/(kg
•
min)的速度收集渗漉液,抽滤,并随时补足75%乙醇,得到总的渗漉液,总的渗漉液的体积量以l计为鬼针草重量以kg计的15倍;取渗漉液,47℃旋转蒸发仪浓缩至无乙醇,得流浸膏,流浸膏的体积量与鬼针草的重量之比为1l:1.5kg,经石油醚、乙酸乙酯、正丁醇分别萃取,萃取至颜色变浅后更换溶剂萃取,萃取完成后,减压回收溶剂,得鬼针草各萃取部位,取乙酸乙酯部位浸膏为萃取浸膏。
[0040]
取萃取浸膏(236.31g),配制成25%乙醇溶液,溶液于离心机内4000r/min离心5分钟,取上清液湿法分次上样于5kg hp
‑
20大孔吸附树脂柱,每次上样量为500ml,上样后,待样品溶液全部进入柱材料,分别以20%
‑
40%
‑
60%
‑
80%
‑
100%乙醇进行梯度洗脱,每个梯度均洗脱至流出液无颜色之后更换下一个梯度;相同的洗脱剂洗脱下来的流分合并,减压回收溶剂后得到相应的浸膏,更换梯度后的流分合并进入下一个梯度,共收集到5个流分段;取40%乙醇流分作为洗脱浸膏;取洗脱浸膏(101.5g),将其配制成12%乙醇溶液,溶液于离心机内4000r/min离心5分钟,取上清液分次湿法上样于3kg聚酰胺柱,每次上样量200ml,上样后,待样品溶液全部流入聚酰胺,以1倍柱体积的水进行冲洗之后,再分别以10%乙醇
‑
50%乙醇
‑
95%乙醇进行洗脱,同样每个梯度均洗脱至流出液基本无颜色后更换下一个梯度;相同的洗脱剂洗脱下来的流分合并,收集共得到3个流分段;取10%乙醇流分作为第二洗脱液;取第二洗脱液,将其配制成40%甲醇溶液,同上述方法一样,将约35g的样品分次上样于小孔吸附树脂(mci)柱1kg,每次上样量50ml;之后分别以35%甲醇
‑
50%甲醇
‑
65%甲醇进行梯度洗脱,分别得到这三个流分段;取50%甲醇流分作为第三洗脱液;继续把第三洗脱液以葡聚糖凝胶sephadexlh20进行分离,以85%甲醇进行洗脱,以自动流分收集器进行收集,每管5ml,收集好后根据聚酰胺薄层色谱板的展开结果进行合并,分别得到a、b、c、d、e5个流分段;取c部位作为第四洗脱液;取第四洗脱液,于47℃旋蒸避光适当浓缩后,通过制备高效液相系统,制备分离出
鬼针草中活性单体成分,所需色谱条件为甲醇:水=33:67,而后所得样品于分析液相210nm处初步检识化合物的纯度,待其纯度达到95%以上后,低温避光旋蒸蒸干溶剂,得干燥粉末。
[0041]
通过质谱(ms)、核磁共振碳谱和氢谱(nmr)、高效液相色谱(hplc)以及查阅既往研究文献等分析方法和手段,对分离所得的化合物进行结构鉴定,结果2个化合物1、2分别鉴定为鬼针聚炔苷b(bipinnata polyacetyloside b)、鬼针聚炔苷(bipinnata polyacetyloside)。
[0042]
鬼针聚炔苷b的收率为0.245
‰
,鬼针聚炔苷收率为0.159
‰
。
[0043]
对比例1与实施例1不同的是:取鬼针草,用粉碎机反复粉碎至全部过2号筛(药典粗粉),共计13.2kg,采用体积分数为95%的乙醇回流提取3次,合并提取液,45℃旋转蒸发仪浓缩至无乙醇,得流浸膏,流浸膏的体积量与鬼针草的重量之比为1l:1kg,经石油醚、乙酸乙酯、正丁醇分别萃取,萃取至颜色变浅后更换溶剂萃取,萃取完成后,减压回收溶剂,得鬼针草各萃取部位,取乙酸乙酯部位浸膏为萃取浸膏。
[0044]
本工艺所得鬼针聚炔苷b的收率为0.0314
‰
,鬼针聚炔苷收率为0.0127
‰
。
[0045]
对比例2本对比例参照如下文献(马明.婆婆针有效部位化学成分的研究[d].山东中医药大学,2003.)记载的提取分离方法进行,具体参阅第3
‑
4页。
[0046]
取鬼针草药材13.2kg,粉碎,用体积浓度为95%的工业乙醇回流提取3次,合并提取液,浓缩,去除叶绿素,硅胶拌样,氯仿
‑
甲醇梯度洗脱;氯仿
‑
甲醇(10:2)洗脱部分拌入聚酰胺,用不同浓度乙醇洗脱,水洗较后部分进行高效液相制备,得到鬼针聚炔苷b (157.73mg),鬼针聚炔苷(100.52mg)。
[0047]
本工艺所得鬼针聚炔苷b的收率为0.0119
‰
,鬼针聚炔苷收率为0.0076
‰
。
[0048]
综上所述,本发明实施例制备的鬼针聚炔苷b和鬼针聚炔苷的收率更高,能够高效、环保地从鬼针草中提取出鬼针聚炔苷b和鬼针聚炔苷,而且这两种化合物被证明具有抗微生物、细胞毒以及抗肿瘤等良好生物活性,能够被应用于制备抗生物毒性药物等。
[0049]
最后说明的是,以上实施例仅用以说明本发明的技术方案而非限制,本领域普通技术人员对本发明的技术方案所做的其他修改或者等同替换,只要不脱离本发明技术方案的精神和范围,均应涵盖在本发明的权利要求范围当中。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1