一株生产N-乙酰神经氨酸的基因工程菌及其构建与应用
一株生产n
‑
乙酰神经氨酸的基因工程菌及其构建与应用
技术领域:
1.本发明属于基因工程技术领域,具体涉及一种利用木糖诱导生产n
‑
乙酰神经氨酸的基因工程菌及其构建与应用。
背景技术:
2.神经氨酸又称唾液酸,是一类含有九个碳原子具有吡喃糖结构的酸性氨基糖。n
‑
乙酰神经氨酸(n
‑
acetyl neuraminic acid,neu5ac)是唾液酸家族中最重要的一员,在生命活动过程中具有重要意义。
3.n
‑
乙酰神经氨酸(neu5ac)通常存在于糖脂和糖蛋白的非还原端,在受精、免疫、分化和神经事件中作为配体受体和细胞
‑
细胞相互作用。n
‑
乙酰神经氨酸具有许多重要的功能,包括提高学习记忆能力,促进婴儿骨骼生长,吸收维生素和矿物质,促进大脑和骨骼发育。neu5ac可进入胎儿和婴儿的神经系统,与糖磷脂结合形成神经节苷脂,促进婴儿神经系统的发育。neu5ac在抗粘连、抗病毒、抗癌等方面具有重要的医学应用价值,是抗病毒药物扎那米韦和奥司他韦的中间体。
4.目前,neu5ac的生产方法主要有天然物质提取、聚合物水解、化学和酶催化、全细胞催化、微生物发酵合成等。与其它方法相比,全细胞催化法不需要额外的atp,节省了生产成本,但大多数反应仍需添加昂贵且过量的丙酮酸和n
‑
乙酰葡萄糖胺作为底物。在整个细胞催化过程中,底物和产物的转移通常受到细胞膜的阻碍,该方法降低了反应速度和产物收率,n
‑
乙酰葡萄糖胺的产率小于60%。这些问题极大的限制了工业上利用全细胞催化法生产neu5ac的发展。
5.因此微生物发酵法逐渐成为了生产neu5ac的热点方向。微生物发酵法利用简单廉价的碳源(如葡萄糖和甘油)直接微生物发酵neu5ac,不需要任何直接的前体和辅助因子,具有广阔的应用前景和市场价值。
6.tian等人利用枯草芽孢杆菌(bacillus subtilis)作为生产菌株,摇瓶发酵得到neu5ac产量为9.71g/l。pang等人利用大肠杆菌(e.coli)作为生产菌株,摇瓶发酵120h得到neu5ac产量为14.23g/l,但生产强度仅有0.12g/(l
×
h),并且没有发酵规模放大的结果报道。以上产量均为目前报道的不同宿主细胞中的最高产量,但仍然无法满足工业生产需求。
技术实现要素:
7.针对上述存在问题,本技术通过构建n
‑
乙酰神经氨酸大肠杆菌生产菌,对大肠杆菌代谢途径进行合理改造,并通过增强主要代谢途径、阻断分解代谢等代谢工程策略,实现了利用廉价碳源葡萄糖高效快速生产neu5ac。本发明提供一种利用木糖诱导生产n
‑
乙酰神经氨酸的基因工程菌,及其构建与应用,并制定了相应的发酵过程控制方案,有良好的工业应用前景。
8.本发明提供的技术方案之一,是一株生产neu5ac的大肠杆菌基因工程菌,所述基因工程菌以大肠杆菌为宿主,具有木糖诱导型启动子p
xylf
控制的来源于t7噬菌体的rna聚
合酶,可通过木糖诱导激活该强表达系统,实现对靶基因的可控强化;糖代谢转录抑制因子突变体mlc*基因替换原有mlc基因;在此基础上敲除了naga、nagb、nagc、nage、manx、many、manz基因,阻断了glcnac的分解代谢途径;在基因组上单拷贝氨基葡萄糖
‑6‑
磷酸n
‑
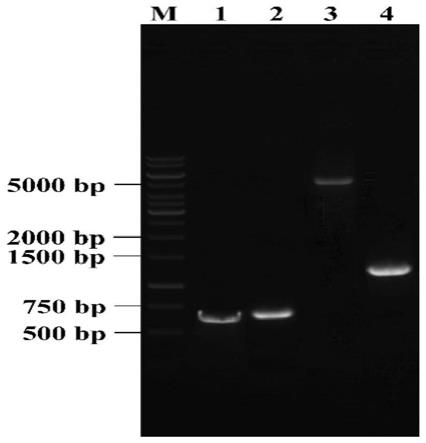
乙酰转移酶基因sc
‑
gna1,双拷贝果糖
‑6‑
磷酸氨基转移酶基因glms,单拷贝磷酸转移酶基因yqab,强化合成neu5ac所需前体物mannac;单拷贝n
‑
乙酰葡萄糖胺2
‑
差向异构酶基因bage,双拷贝n
‑
乙酰神经氨酸合成酶基因neub,构建neu5ac合成通路;并敲除neu5ac分解代谢相关基因nanatek,实现了大肠杆菌中neu5ac的从头合成;敲除pts系统相关基因ptsg,消除葡萄糖特异性pts系统,并整合葡萄糖易化转运蛋白基因glf、大肠杆菌自身葡萄糖激酶基因glk,强化合成neu5ac所需前体物磷酸烯醇式丙酮酸的供应;在此基础上,敲除丙酮酸激酶pyka,进一步强化磷酸烯醇式丙酮酸的积累。
9.所述t7 rna聚合酶基因,核苷酸序列如序列表seq id no.1所示;
10.所述氨基葡萄糖
‑6‑
磷酸n
‑
乙酰转移酶基因sc
‑
gna1源自酿酒酵母(saccharomyces cerevisiae),核苷酸序列如序列表seq id no.2所示,由p
t7
启动子控制;
11.所述果糖
‑6‑
磷酸转氨酶基因glms源自大肠杆菌(e.coli)w3110,核苷酸序列如序列表seq id no.3所示,由p
t7
启动子控制;
12.所述n
‑
乙酰氨基葡萄糖2
‑
差向异构酶基因bage源自项圈藻(anabaena sp.)ch1,核苷酸序列如序列表seq id no.4所示,p
t7
启动子控制;
13.所述n
‑
乙酰神经氨酸合成酶基因neub源自脑膜炎奈瑟菌(meningococcus),核苷酸序列如序列表seq id no.5所示,由p
t7
启动子控制;
14.所述磷酸转移酶基因yqab源自大肠杆菌(e.coli)w3110,核苷酸序列如序列表seq id no.6所示,由p
trc
启动子控制;
15.所述葡萄糖易化转运蛋白基因glf源自运动发酵假单胞菌(zymomonas mobilis),核苷酸序列如序列表seq id no.7所示,由p
trc
启动子控制;
16.所述葡萄糖激酶基因glk源自大肠杆菌(e.coli)w3110,核苷酸序列核苷酸序列如序列表seq id no.8所示,由p
trc
启动子控制;
17.所述糖代谢转录抑制因子突变基因mlc*,核苷酸序列如序列表seq id no.9所示;
18.优选地,所述基因工程菌以大肠杆菌e.coli w3110为出发菌株。
19.本发明提供的技术方案之二,是上述基因工程菌的构建方法,对e.coli w3110进行基因组水平定向改造,具体包括如下步骤:
20.(1)在laciz(gene id:945007,945006)基因位点上整合由木糖启动子p
xylf
控制的t7rna聚合酶,引入糖代谢转录抑制因子突变基因mlc*替换原有mlc基因,解除葡萄糖抑制作用。
21.(2)构建glcnac合成通路。首先敲除glcnac的分解代谢途径相关基因:naga(gene id:945289)、nagb(gene id:945290),nagc(gene id:945285)、nage(gene id:945292)、manx(gene id:946334)、many(gene id:946332)、manz(gene id:946342),同时在nage(gene id:945292)基因位点整合由p
t7
启动子控制的氨基葡萄糖
‑6‑
磷酸n
‑
乙酰转移酶基因sc
‑
gna1;在假基因位点yjiv(gene id:2847669)和假基因位点ycjv(gene id:945890)整合由p
t7
启动子控制的果糖
‑6‑
磷酸转氨酶基因glms。
22.(3)在构建上述glcnac合成通路的基础上,在假基因位点ilvg(gene id:2847699)
整合p
trc
控制的磷酸转移酶基因yqab,进一步促进前体物n
‑
乙酰葡萄糖胺在胞内的合成。
23.(4)为了构建合成通路,敲除了neu5ac的分解代谢途径nana(gene id:947742)、nant(gene id:947740)、nane(gene id:947745)、nank(gene id:947757),并在假基因位点gapc(gene id:2847738)整合由p
t7
启动子控制的n
‑
乙酰葡萄糖胺2
‑
差向异构酶基因bage,在假基因位点yeep(gene id:946524)和假基因位点mbha(gene id:944921)上分别整合了由p
t7
启动子控制的n
‑
乙酰神经氨酸合成酶基因neub。
24.(5)为了加强前体物质磷酸烯醇式丙酮酸的积累,首先敲除pts系统相关基因ptsg(gene id:945651),在假基因位点rph(gene id:948156)上整合由p
trc
启动子控制glf,在假基因位点ycgh(gene id:2847703)整合由p
trc
启动子控制glk;在此基础上敲除丙酮酸激酶pyka(gene id:946527)。
25.上述构建顺序不分先后,可根据需求进行调整。
26.本发明还提供上述基因工程菌在生产neu5ac中的应用;
27.进一步地,利用上述基因工程菌发酵生产neu5ac的方法如下:
28.1、摇瓶发酵
29.(1)活化斜面培养:用接种环从
‑
80℃冰箱保菌管中接种1
‑
2环菌种,均匀涂布于斜面培养基中,35
‑
39℃培养8
‑
16h,转接到第二代斜面培养基中,35
‑
39℃培养8
‑
16h;
30.(2)种子瓶培养:用接种环将斜面上的菌体接种到装有30ml种子培养基的500ml三角瓶中用于制备种子液,三角瓶使用十二层纱布封口,在35
‑
39℃,180
‑
240r/min的条件下振荡培养8
‑
16h;
31.(3)发酵培养:将种子液按10
‑
15%接种量接种到发酵培养基中,使其终体积为30ml,使用十二层纱布封口,在35
‑
39℃,180
‑
240r/min的条件下振荡培养,发酵过程中通过补加氨水维持ph在6.8
‑
7.2,当通过苯酚红指示剂观察到ph没有缓慢降低甚至有所升高,表明菌体缺糖,补加0.5
‑
2ml 60%(m/v)葡萄糖溶液,发酵周期为30
‑
36h,产量达到10.5
‑
11.36g/l,生产强度可达0.35~0.38g/(l
×
h);
32.2、发酵罐发酵
33.(1)斜面活化培养:取出于
‑
80℃保存的微生物菌种,在无菌条件下密集划线接种至斜面培养基,置于36℃~38℃培养约12~14h,培养完毕后转接于斜面茄形瓶活化培养基传代培养一次;
34.(2)种子罐培养:将200ml左右无菌水于超净台火焰附近倒入茄形瓶中,使用接种环将菌落刮至无菌水中制备菌悬液,将菌悬液接种入种子培养基中,进行种子培养,培养过程ph维持在7.0
‑
7.2左右,温度维持在35
‑
39℃左右,溶氧维持在25
‑
40%之间,培养至细胞干重3
‑
5g/l转接发酵罐发酵培养;
35.(3)发酵罐发酵培养:按照10
‑
20%的接种量将种子液接种至发酵培养基进行发酵,发酵过程中ph维持在7.0
‑
7.2左右,温度维持在35
‑
39℃左右,溶氧维持在25
‑
40%之间,培养过程中通过流加60
‑
80%(m/v)葡萄糖溶液维持发酵过程,并保持发酵过程中培养液中葡萄糖浓度维持在0.1
‑
5g/l之间,发酵周期约36
‑
50h;
36.经过36
‑
50h的发酵,neu5ac的发酵液产量达到25~28g/l,生产强度可达0.64~0.67g/(l
×
h)。
37.优选的,所述斜面培养基组成为:酵母粉3
‑
8g/l、蛋白胨5
‑
15g/l、nacl 3
‑
10g/l、
牛肉膏5
‑
15g/l、蔗糖0.5
‑
2g/l、琼脂粉15
‑
30g/l,其余为水,ph 6.8
‑
7.2。
38.优选的,所述种子培养基组成为:葡萄糖15
‑
25g/l,酵母粉2
‑
5g/l,(nh4)2so
4 1
‑
5g/l,kh2po
4 1
‑
5g/l,mgso4·
7h2o 0.5
‑
2.5g/l,feso4·
7h2o 1
‑
5mg/l,mnso4·
7h2o 1
‑
5mg/l,v
h 0.05
‑
5mg/l,v
b1 0.1
‑
2mg/l,微量元素混合液1
‑
3ml/l,消泡剂1
‑
2滴,其余为水,ph 6.8
‑
7.2。
39.优选的,所述发酵培养基组成为:葡萄糖15
‑
30g/l,木糖5
‑
20g/l,酵母粉2
‑
5g/l,(nh4)2so
42‑
10g/l,kh2po44
‑
10g/l,mgso4·
7h2o 2
‑
8g/l,nacl 0.5
‑
3g/l,feso4·
7h2o 5
‑
30mg/l,mnso4
·
7h2o 1
‑
5mg/l,cacl2·
2h2o 15
‑
30mg/l,v
h 0.05
‑
2mg/l,v
b1 0.1
‑
1mg/l,微量元素混合液1
‑
3ml/l,苯酚红指示剂1
‑
3%,消泡剂1
‑
2滴,其余为水,ph 6.8
‑
7.2。
40.优选的,微量元素混合液组成为:na2moo4·
2h2o 1
‑
3g/l,nicl2·
6h2o 0.5
‑
1.5g/l,cacl2·
2h2o 2
‑
8g/l,cuso4·
5h2o 0.1
‑
0.5g/l,al2(so4)3·
18h2o 1
‑
1.5g/l,cocl2·
6h2o 0.5
‑
1.5g/l,znso4·
2h2o 0.1
‑
0.5g/l,h3bo30.05
‑
0.2g/l,其余为水。
41.有益效果:
42.本发明以e.coli w3110为出发菌株,在基因组上整合n
‑
乙酰氨基葡萄糖合成途径,并引入来自项圈藻的n
‑
乙酰氨基葡萄糖2
‑
差向异构酶基因bage以及来自脑膜炎奈瑟菌的n
‑
乙酰神经氨酸合成酶基因neub,构建n
‑
乙酰神经氨酸合成途径,并敲除其分解代谢途径的关键基因nanatek。同时对合成n
‑
乙酰神经氨酸所需的前体物质的代谢途径进行多拷贝强化并对部分旁路代谢途径进行敲除,通过对产glcnac和neu5ac的关键酶基因进行不同拷贝数的优化,最终确定了关键酶基因的最佳比例,得到一株n
‑
乙酰神经氨酸高产菌。5l发酵罐发酵42h,n
‑
乙酰神经氨酸产量最高可达28g/l,生产强度最高可达0.67g/(l
×
h),为目前报道的最高值,具有重要的工业应用价值。
附图说明:
43.图1:nanatek基因敲除片段构建及电泳验证图。其中:m:1kb dna marker;1:上游同源臂;2:下游同源臂;3:原菌对照;4:阳性菌鉴定片段。
44.图2:ptsg基因敲除片段构建及电泳验证图。其中:m:1kb dna marker;1:上游同源臂;2:下游同源臂;3:原菌对照;4:阳性菌鉴定片段。
45.图3:pyka基因敲除片段构建及电泳验证图。其中:m:1kb dna marker;1:上游同源臂;2:下游同源臂;3:原菌对照;4:阳性菌鉴定片段。
46.图4:yeep位点整合neub基因整合片段构建及电泳验证图。其中:m:1kb dna marker;1:上游同源臂;2:下游同源臂;3:p
t7
‑
neub基因片段;4:原菌对照;5:阳性菌鉴定片段。
47.图5:gapc位点bage基因整合片段构建及电泳验证图。其中:m:1kb dna marker;1:上游同源臂;2:下游同源臂;3:p
t7
‑
bage基因片段;4:原菌对照;5:阳性菌鉴定片段。
48.图6:rph位点整合glf基因整合片段构建及电泳验证图。其中:m:1kb dna marker;1:上游同源臂;2:下游同源臂;3:p
trc
‑
glf基因片段;4:原菌对照;5:阳性菌鉴定片段。
49.图7:ycgh位点整合glk基因整合片段构建及电泳验证图。其中:m:1kb dna marker;1:上游同源臂;2:下游同源臂;3:p
trc
‑
glk基因片段;4:原菌对照;5:阳性菌鉴定片段。
具体实施方式:
50.下面通过具体的实施方案叙述本发明。除非特别说明,本发明中所用的试验方法均为本领域技术人员所公知的方法。另外,实施方案应理解为说明性的,而非限制本发明的范围,本发明的实质和范围仅由权利要求书所限定。对于本领域技术人员而言,在不背离本发明实质和范围的前提下,对这些实施方案中的物料成分和用量进行的各种改变或改动也属于本发明的保护范围。
51.实施例1:neu5ac生产大肠杆菌基因工程菌的构建
52.采用crispr/cas9基因编辑技术对基因进行定向改造。本发明中采用的基因编辑方法参照文献(li y,lin z,huang c,et al.metabolic engineering of escherichia coli using crispr
–
cas9 meditated genome editing.metabolic engineering,2015,31:13
‑
21.)进行。
53.涉及到的部分实验方法如下:
54.(1)pgrb质粒的构建:
55.使用crispr rgen tools设计靶序列(pam:5
’‑
ngg
‑3’
)用于切割靶基因。合成正向引物和反向互补引物后,各取10μl于pcr管中,混合均匀后通过单链dna的退火制备包含靶序列的dna片段。反应条件:预变性95℃,5min;退火50℃,1min。将得到的dna片段与通过反向pcr获得的线性化pgrb载体,通过同源重组连接,得到pgrb质粒。同源重组所用试剂盒为ⅱonestepcloningkit系列。
56.(2)重组dna片段的构建:
57.敲除目的基因所需重组dna片段为目的基因上游同源臂和下游同源臂两个片段重叠而成。整合目的基因所需重组片段为整合位点基因的上、下游同源臂和目的基因三个片段重叠而成。
58.以待敲除基因或整合目的基因的待整合位点的上、下游序列为模板,设计上、下游同源臂引物,通常使同源臂长度在500bp左右,以待整合基因为模板,设计整合基因的扩增引物。通过pcr的方法分别扩增上下游同源臂和目的基因片段后,再经重叠pcr制备重组片段。
59.(3)感受态细胞的制备:
60.在37℃,220rpm条件下,用装有100ml2
×
yt培养基的三角瓶培养菌体至od
600
=0.4
‑
0.6时进行感受态制备。当细胞内携带predcas9质粒时,培养温度调整至32℃,且当菌体od
600
=0.1
‑
0.2时添加0.1m的iptg。制备过程参照常规标准操作。
61.(4)pgrb质粒和重组dna的转化:
62.将pgrb质粒和重组dna片段同时电转化至含有predcas9的电转感受态细胞中。将经过电转化的菌体复苏培养2h后涂布于含氨苄青霉素和奇霉素的lb平板上,32℃过夜培养。用上游同源臂的上游引物和下游同源臂的下游引物,或设计专门的鉴定引物,进行菌落pcr验证,筛选阳性重组子。
63.(5)质粒的消除:
64.将阳性重组子置于含有0.2%阿拉伯糖的lb培养基中过夜培养后涂布于含有奇霉素抗性的lb平板上,再次32℃过夜培养。挑选单菌落对点含有氨苄青霉素和奇霉素抗性的lb平板,挑选氨苄青霉素平板不生长,奇霉素抗性平板生长的单菌落即为消除pgrb质粒的
重组菌株。将阳性重组子转接到无抗性的lb液体培养基中,42℃过夜培养后涂布于无抗性的lb平板上,37℃过夜培养。挑选单菌落对点含有奇霉素抗性和无抗性的lb平板,挑选奇霉素抗性平板不生长,无抗性平板生长的单菌落即为消除predcas9质粒的重组菌株。
65.(6)本发明所述用的启动子
66.p
t7
启动子,核苷酸序列如下所示:taatacgact cactataggg tctagaaata attttgttta actttaagaa ggagatatacc;
67.p
trc
启动子,核苷酸序列如下所示:ttgacaattaatcatccggctcgtataatgtgtggaattgtgagcggataacaatttcacacaggaaacagacc;
68.p
xylf
启动子,核苷酸序列如下所示:ctagcataac cccttggggc ctctaaacgg gtcttgaggg gttttttg。
69.菌株构建的具体方法如下:
70.(1)前体物glcnac合成通路的构建
71.以大肠杆菌(e.coli w3100)为出发菌株,在基因位点laciz上整合p
xylf
‑
t7rnap,用文献报道的突变基因mlc*替换mlc基因,并进一步进行glcnac合成通路的构建。
72.以大肠杆菌(e.coli w3110)基因组为模板,根据其nagbac基因簇的上、下游序列设计上游同源臂引物nagbac
‑
f1、nagbac
‑
r1和下游同源臂引物nagbac
‑
f2、nagbac
‑
r2,先通过pcr得到其上、下游同源臂,再通过重叠pcr的方法获得重叠片段(nagbac
‑
u—nagbac
‑
d)。通过grna搜索工具(http://www.rgenome.net/cas
‑
designer)查找合适的grna序列,进行序列合成后,将其与pgrb线性化载体同源重组得到pgrb
‑
nagbac。将重叠片段和pgrb
‑
nagbac电转化至含有predcas9载体的e.coli w3110感受态细胞中,将电转化后复苏培养的菌体涂布于含氨苄青霉素和奇霉素的lb平板上,32℃过夜培养后利用菌落pcr验证阳性重组子,再消除用于基因编辑的pgrb
‑
nagbac,最终获得成功敲除nagbac基因的阳性菌株。
73.以同样的方法继续敲除manxyz基因,阻断glcnac的分解途径。
74.用文献报道的突变基因mlc*替换mlc基因,先进行mlc基因的敲除,再在敲除位点附近进行mlc*基因的整合。具体步骤如下:
75.以大肠杆菌(e.coli w3110)基因组为模板,根据其mlc基因的上、下游序列设计上游同源臂引物mlc
‑
f1、mlc
‑
r1和下游同源臂引物mlc
‑
f2、mlc
‑
r2(在mlc
‑
r1和mlc
‑
f2中引入后续利用crispr/cas9技术进行基因整合的切割位点),先通过pcr得到其上、下游同源臂,再通过重叠pcr的方法获得重叠片段(mlc
‑
u—mlc
‑
d)。通过grna搜索工具(http://www.rgenome.net/cas
‑
designer)查找合适的grna序列,进行序列合成后,将其与pgrb线性化载体同源重组得到pgrb
‑
mlc。将重叠片段和pgrb
‑
mlc电转化至含有predcas9载体的e.coli w3110感受态细胞中,将电转化后复苏培养的菌体涂布于含氨苄青霉素和奇霉素的lb平板上,32℃过夜培养后利用菌落pcr验证阳性重组子,再消除用于基因编辑的pgrb
‑
mlc,最终获得成功敲除mlc基因的阳性菌株。
76.进一步以大肠杆菌(e.coli w3110)基因组为模板,根据原始mlc基因位点附近的上、下游序列设计上游同源臂引物mlc*
‑
f1、mlc*
‑
r1和下游同源臂引物mlc*
‑
f2、mlc*
‑
r2,以大肠杆菌(e.coli w3110)基因组为模板,根据mlc*基因的序列设计引物。先通过pcr得到其上下游同源臂(mlc*
‑
u、mlc*
‑
d)及突变的mlc*基因,再通过重叠pcr的方法获得重叠片段(mlc*
‑
u—mlc*—mlc*
‑
d)。根据mlc
‑
r1和mlc
‑
f2中引入的切割位点设计grna序列(间隔区
‑
pam序列为tgcgctggttgatttcttctagg),进行序列合成后,将其与pgrb线性化载体同源重组得到pgrb
‑
mlc*。将重叠片段和pgrb
‑
mlc*电转化至含有predcas9载体的上步构建的阳性菌株感受态细胞中,将电转化后复苏培养的菌体涂布于含氨苄青霉素和奇霉素的lb平板上,32℃过夜培养后利用菌落pcr验证阳性重组子,再消除用于基因编辑的pgrb
‑
mlc*,最终获得成功敲除mlc基因并整合mlc*基因的阳性转菌株。以大肠杆菌(e.coli w3110)基因组为模板,根据其nage基因簇的上、下游序列设计上游同源臂引物nage
‑
f1、nage
‑
r1和下游同源臂引物nage
‑
f2、nage
‑
r2,以酿酒酵母(saccharomyces cerevisiae)基因组为模板,根据其sc
‑
gna1基因的序列设计引物,并用p
t7
启动子控制sc
‑
gna1的表达。先通过pcr得到其上下游同源臂(nage
‑
u、nage
‑
d)及目的基因p
t7
控制的sc
‑
gna1基因(p
t7
‑
gna1),再通过重叠pcr的方法获得重叠片段(nage
‑
u—p
t7
‑
gna1—nage
‑
d)。通过grna搜索工具(http://www.rgenome.net/cas
‑
designer)查找合适的grna序列,进行序列合成后,将其与pgrb线性化载体同源重组得到pgrb
‑
nage。将重叠片段和pgrb
‑
nage电转化至含有predcas9载体的上步构建的阳性菌株感受态细胞中,将电转化后复苏培养的菌体涂布于含氨苄青霉素和奇霉素的lb平板上,32℃过夜培养后利用菌落pcr验证阳性重组子,再消除用于基因编辑的pgrb
‑
nage,最终获得成功敲除nage基因并整合p
t7
‑
gna1基因的阳性菌株。
77.在基因位点laciz上整合p
xylf
‑
t7rnap(p
xylf
控制的t7rnap)p
xylf
‑
t7rnap,在假基因位点yjiv和假基因位点ycjv上分别整合p
t7
‑
glms(p
t7
控制的glms基因)基因的方法均同上,以进一步强化glcnac合成通路。
78.(2)neu5ac合成通路的构建
79.以大肠杆菌(e.coli w3110)基因组为模板,根据其nanatek基因的上、下游序列设计上游同源臂引物nanatek
‑
f1、nanatek
‑
r1和下游同源臂引物nanatek
‑
f2、nanatek
‑
r2,先通过pcr得到其上下游同源臂,再通过重叠pcr的方法获得重叠片段(nanatek
‑
u—nanatek
‑
d)。通过grna搜索工具(http://www.rgenome.net/cas
‑
designer)查找合适的grna序列,合成grna
‑
nanatek
‑
s与grna
‑
nanatek
‑
a序列,通过pcr退火将两条单链引物互补配对以得到双链grna
‑
nanatek,将其与pgrb线性化载体同源重组得到pgrb
‑
nanatek。将重叠片段和pgrb
‑
nanatek电转化至含有predcas9载体的步骤(1)获得的阳性转化子感受态细胞中,将电转化后复苏培养的菌体涂布于含氨苄青霉素和奇霉素的lb平板上,32℃过夜培养后利用菌落pcr验证阳性重组子,再消除用于基因编辑的pgrb
‑
nanatek,最终获得成功敲除nanatek的阳性菌株。
80.敲除nanatek基因重叠片段的构建和阳性菌株的pcr验证的电泳图见附图1。其中:m:1kb dna marker1:上游同源臂602bp;2:下游同源臂636bp;3:原菌对照5046bp;4:阳性菌鉴定片段1238bp。
81.以同样的操作敲除ptsg、pyka基因。消除葡萄糖特异性pts系统、前体物磷酸烯醇式丙酮酸的分解途径。
82.敲除ptsg基因重叠片段的构建和阳性菌株的pcr验证的电泳图见附图2。其中:m:1kb dna marker1:上游同源臂629bp;2:下游同源臂450bp;3:原菌对照2056bp;4:阳性菌鉴定片段1079bp。
83.敲除pyka基因重叠片段的构建和阳性菌株的pcr验证的电泳图见附图3。其中:m:1kb dna marker1:上游同源臂608bp;2:下游同源臂457bp;3:原菌对照2382bp;4:阳性菌鉴
定片段1065bp。
84.以大肠杆菌(e.coli w3110)基因组为模板,根据其yeep基因的上、下游序列设计上游同源臂引物yeep
‑
f1、yeep
‑
r1和下游同源臂引物yeep
‑
f2、yeep
‑
r2,以大肠杆菌(e.coli w3110)基因组为模板,根据其neub基因的序列设计引物,启动子p
trc
则设计在yeep基因上游同源臂的下游引物和基因的上游引物中,先通过pcr得到yeep基因上下游同源臂(yeep
‑
u、yeep
‑
d)及目的基因(p
t7
‑
neub,p
t7
控制的neub基因),再通过重叠pcr的方法获得重叠片段(yeep
‑
u—p
t7
‑
neub—yeep
‑
d)。通过grna搜索工具(http://www.rgenome.net/cas
‑
designer)查找合适的grna序列,合成grna
‑
yeep
‑
s与grna
‑
yeep
‑
a序列,通过pcr退火将两条单链引物互补配对以得到双链grna
‑
yeep,将其与pgrb线性化载体同源重组得到pgrb
‑
yeep。将重叠片段和pgrb
‑
yeep电转化至含有predcas9载体的上步构建的阳性转化子感受态细胞中,将电转化后复苏培养的菌体涂布于含氨苄青霉素和奇霉素的lb平板上,32℃过夜培养后利用菌落pcr验证阳性重组子,再消除用于基因编辑的pgrb
‑
yeep,最终获得成功敲除yeep基因并整合p
t7
‑
neub的阳性菌株。
85.yeep位点整合p
t7
‑
neub基因重叠片段的构建和阳性菌株的pcr验证的电泳图见附图4。其中:m:1kb dna marker;1:上游同源臂558bp;2:下游同源臂547bp;3:目的基因1130bp;4:原菌对照1396bp;5:阳性菌鉴定片段2184bp。
86.以同样的方法在假基因位点gapc上整合p
t7
‑
bage(p
t7
控制的bage基因),在假基因位点mbha上整合p
t7
‑
neub(p
t7
控制的neub基因),在假基因位点ilvg上整合p
trc
‑
yqab,在假基因位点rph上整合p
trc
‑
glf(p
trc
控制的glf基因),在假基因位点ycgh上整合p
trc
‑
glk(p
trc
控制的glk基因)。构建并强化neu5ac合成通路,最终获得neu5ac生产菌株。
87.gapc位点整合p
t7
‑
bage基因重叠片段的构建和阳性菌株的pcr验证的电泳图见附图5。其中:m:1kb dna marker;1:上游同源臂476bp;2:下游同源臂505bp;3:目的基因1196bp;4:原菌对照1681bp;5:阳性菌鉴定片段2126bp。
88.rph位点整合p
trc
‑
glf基因重叠片段的构建和阳性菌株的pcr验证的电泳图见附图6。其中:m:1kb dna marker;1:上游同源臂523bp;2:下游同源臂550bp;3:目的基因1545bp;4:原菌对照1359bp;5:阳性菌鉴定片段2537bp。
89.ycgh位点整合p
trc
‑
glk基因重叠片段的构建和阳性菌株的pcr验证的电泳图见附图7。其中:m:1kb dna marker;1:上游同源臂573bp;2:下游同源臂630bp;3:目的基因1089bp;4:原菌对照1409bp;5:阳性菌鉴定片段2211bp。
90.上述构建顺序不分先后,可根据需求进行调整。最终获得具有木糖诱导型启动子p
xylf
控制的来源于t7噬菌体的rna聚合酶,糖代谢转录抑制因子突变体mlc*基因替换原有mlc基因,敲除了naga、nagb、nagc、nage、manx、many、manz基因,在基因组上单拷贝氨基葡萄糖
‑6‑
磷酸n
‑
乙酰转移酶基因sc
‑
gna1,双拷贝果糖
‑6‑
磷酸氨基转移酶基因glms,单拷贝磷酸转移酶基因yqab,单拷贝n
‑
乙酰葡萄糖胺2
‑
差向异构酶基因bage,双拷贝n
‑
乙酰神经氨酸合成酶基因neub,敲除neu5ac分解代谢相关基因nanatek,敲除pts系统相关基因ptsg,整合葡萄糖易化转运蛋白基因glf、葡萄糖激酶基因glk,敲除丙酮酸激酶pyka的生产neu5ac的大肠杆菌基因工程菌。
91.表1菌株构建过程中所涉及的引物
92.93.94.[0095][0096]
实施例2:利用上述实施例1所构建的菌株作为生产菌株生产neu5ac的方法
[0097]
(1)斜面活化培养:取出于
‑
80℃保存的微生物菌种,在无菌条件下密集划线接种至斜面活化培养基,置于37℃培养约12h,培养完毕后转接于斜面茄形瓶活化培养基传代培养一次;
[0098]
(2)种子罐培养:将200ml左右无菌水于超净台火焰附近倒入茄形瓶中,使用接种环将菌落刮至无菌水中制备菌悬液,将菌悬液接种入种子培养基中,进行种子培养;培养过程ph维持在7.0,温度维持在37℃,溶氧维持在30%,培养至细胞干重3g/l转接发酵罐发酵培养;
[0099]
(3)发酵罐发酵培养:按15%的接种量将种子液接种至发酵培养基进行发酵,发酵过程中ph维持在7.0,温度维持在37℃,溶氧维持在30%,培养过程中通过流加80%(m/v)葡萄糖溶液维持发酵过程,并保持发酵过程中培养液中葡萄糖浓度维持在1g/l,发酵周期42h,产量达到28g/l,生产强度可达0.67g/(l
×
h)。
[0100]
采用发明专利申请202011432150.1:《木糖诱导生产n
‑
乙酰神经氨酸的工程菌及其应用》中构建的菌株e.coli w3110 neu5ac以上述同样的发酵条件进行发酵罐发酵,发酵周期45h,产量达到12g/l,生产强度可达0.27g/(l
×
h)。
[0101]
本技术中n
‑
乙酰神经氨酸产量提升约133.3%,生产强度提升148.1%。
[0102]
斜面培养基成分为:蛋白胨7g/l,酵母粉4g/l,nacl 8g/l,牛肉膏12g/l,蔗糖1g/l,琼脂粉30g/l,其余为水,ph 6.8
‑
7.2。
[0103]
种子培养基成分为:葡萄糖25g/l,酵母粉5g/l,(nh4)2so41g/l,kh2po45g/l,mgso4·
7h2o 2g/l,feso4·
7h2o 3mg/l,mnso4·
7h2o 5mg/l,v
h 3mg/l,v
b1
1mg/l,微量元素混合液1ml/l,消泡剂2滴,其余为水,ph 6.8
‑
7.2。
[0104]
发酵培养基成分为:葡萄糖15g/l,木糖20g/l,酵母粉4g/l,(nh4)2so
4 8g/l,kh2po
4 4g/l,mgso4·
7h2o 6g/l,nacl 2g/l,feso4·
7h2o 25mg/l,mnso4·
7h2o 4mg/l,cacl2·
2h2o 20mg/l,v
h 2mg/l,v
b1
1mg/l,微量元素混合液2ml/l,苯酚红指示剂1%,消泡剂2滴,其余为水,ph 6.8
‑
7.2。
[0105]
微量元素混合液成分为:na2moo4·
2h2o 2g/l,nicl2·
6h2o 1g/l,cacl2·
2h2o 5g/l,cuso4·
5h2o 0.1g/l,al2(so4)3·
18h2o 1g/l,cocl2·
6h2o 1.5g/l,znso4·
2h2o 0.5g/l,h3bo
3 0.2g/l,其余为水。
[0106]
实施例3:利用上述实施例1所构建的菌株作为生产菌株生产neu5ac的方法
[0107]
(1)斜面活化培养:取出于
‑
80℃保存的微生物菌种,在无菌条件下密集划线接种至斜面活化培养基,置于36℃培养约13h,培养完毕后转接于斜面茄形瓶活化培养基传代培养一次;
[0108]
(2)种子罐培养:将200ml左右无菌水于超净台火焰附近倒入茄形瓶中,使用接种环将菌落刮至无菌水中制备菌悬液,将菌悬液接种入种子培养基中,进行种子培养;培养过程ph维持在7.2,温度维持在36℃,溶氧维持在25%,培养至细胞干重4g/l转接发酵罐发酵培养;
[0109]
(3)发酵罐发酵培养:按10%的接种量将种子液接种至发酵培养基进行发酵,发酵过程中ph维持在7.2,温度维持在36℃,溶氧维持在25%,培养过程中通过流加80%(m/v)葡萄糖溶液维持发酵过程,并保持发酵过程中培养液中葡萄糖浓度维持在1g/l,发酵周期39h,产量达到25g/l,生产强度可达0.64g/(l
×
h)。
[0110]
斜面培养基成分为:蛋白胨12g/l,酵母粉3g/l,nacl 8g/l,牛肉膏15g/l,蔗糖0.8g/l,琼脂粉20g/l,其余为水,ph 6.8
‑
7.2。
[0111]
种子培养基成分为:葡萄糖20g/l,酵母粉5g/l,(nh4)2so
4 3g/l,kh2po
4 3g/l,mgso4·
7h2o 2.5g/l,feso4·
7h2o 3mg/l,mnso4·
7h2o 4mg/l,v
h 0.05mg/l,v
b1 0.5mg/l,微量元素混合液2ml/l,消泡剂2滴,其余为水,ph 6.8
‑
7.2。
[0112]
发酵培养基成分为:葡萄糖25g/l,木糖15g/l,酵母粉5g/l,(nh4)2so
4 7g/l,kh2po
4 10g/l,mgso4·
7h2o 5g/l,nacl 3g/l,feso4·
7h2o 30mg/l,mnso4·
7h2o 3mg/l,cacl2·
2h2o 30mg/l,v
h 0.5mg/l,v
b1
1mg/l,微量元素混合液2ml/l,苯酚红指示剂1%,消泡剂2滴,其余为水,ph 6.8
‑
7.2。
[0113]
微量元素混合液成分为:na2moo4·
2h2o 3g/l,nicl2·
6h2o 1.5g/l,cacl2·
2h2o 8g/l,cuso4·
5h2o 0.5g/l,al2(so4)3·
18h2o 1.5g/l,cocl2·
6h2o 1.5g/l,znso4·
2h2o 0.5g/l,h3bo
3 0.05g/l,其余为水。
[0114]
实施例4:利用上述实施例1所构建的菌株作为生产菌株生产neu5ac的方法
[0115]
(1)斜面活化培养:取出于
‑
80℃保存的微生物菌种,在无菌条件下密集划线接种至斜面活化培养基,置于38℃培养约10h,培养完毕后转接于斜面茄形瓶活化培养基传代培养,38℃培养10h;
[0116]
(2)种子罐培养:将200ml左右无菌水于超净台火焰附近倒入茄形瓶中,使用接种
环将菌落刮至无菌水中制备菌悬液,将菌悬液接种入种子培养基中,进行种子培养;培养过程ph维持在7.1,温度维持在38℃,溶氧维持在40%,培养至细胞干重5g/l转接发酵罐发酵培养;
[0117]
(3)发酵罐发酵培养:按20%的接种量将种子液接种至发酵培养基进行发酵,发酵过程中ph维持在7.1,温度维持在38℃,溶氧维持在40%,培养过程中通过流加80%(m/v)葡萄糖溶液维持发酵过程,并保持发酵过程中培养液中葡萄糖浓度维持在3g/l,发酵周期45h,产量达到26g/l,生产强度可达0.58g/(l
×
h)。
[0118]
斜面培养基成分为:蛋白胨5g/l,酵母粉3g/l,nacl 3g/l,牛肉膏10g/l,蔗糖0.5g/l,琼脂粉20g/l,其余为水,ph 6.8
‑
7.2。
[0119]
种子培养基成分为:葡萄糖15g/l,酵母粉2g/l,(nh4)2so
4 2.5g/l,kh2po
4 2g/l,mgso4·
7h2o 2g/l,feso4·
7h2o 2mg/l,mnso4·
7h2o 3mg/l,v
h 2mg/l,v
b1 1mg/l,微量元素混合液3ml/l,消泡剂2滴,其余为水,ph 6.8
‑
7.2。
[0120]
发酵培养基成分为:葡萄糖15g/l,木糖5g/l,酵母粉2g/l,(nh4)2so
4 2g/l,kh2po
4 4g/l,mgso4·
7h2o 2.5g/l,nacl 2g/l,feso4·
7h2o 20mg/l,mnso4·
7h2o 5mg/l,cacl2·
2h2o 20mg/l,v
h 1mg/l,v
b1 1mg/l,微量元素混合液3ml/l,苯酚红指示剂1%,消泡剂2滴,其余为水,ph 6.8
‑
7.2。
[0121]
微量元素混合液成分为:na2moo4·
2h2o 1g/l,nicl2·
6h2o 0.5g/l,cacl2·
2h2o 2g/l,cuso4·
5h2o 0.1g/l,al2(so4)3·
18h2o 1g/l,cocl2·
6h2o 0.5g/l,znso4·
2h2o 0.1g/l,h3bo
3 0.05g/l,其余为水。
[0122]
实施例5:利用上述实施例1所构建的菌株作为生产菌株生产neu5ac的方法
[0123]
(1)活化斜面培养:
[0124]
用接种环从
‑
80℃冰箱保菌管中接种2环菌种,均匀涂布于斜面培养基中,36℃培养13h,转接到第二代斜面培养基中,36℃培养12h。
[0125]
(2)种子瓶培养:
[0126]
用接种环将斜面上的菌体接种到装有30ml种子培养基的500ml三角瓶中用于制备种子液,三角瓶使用十二层纱布封口,在37℃,220r/min的条件下振荡培养12h。
[0127]
(3)摇瓶发酵:
[0128]
将种子液按10%接种量接种到装有发酵培养基的500ml挡板瓶中,使其终体积为30ml,使用十二层纱布封口,在37℃,220r/min的条件下振荡培养,发酵过程中通过补加氨水维持ph在7.0,当通过苯酚红指示剂观察到ph没有缓慢降低甚至有所升高,表明菌体缺糖,补加1ml 60%(m/v)葡萄糖溶液。
[0129]
斜面培养基成分为:酵母粉7g/l、蛋白胨14g/l、nacl 8g/l、牛肉膏6g/l、蔗糖2g/l、琼脂粉25g/l,其余为水,ph7.0。
[0130]
种子培养基成分为:葡萄糖20g/l,酵母粉4g/l,(nh4)2so
4 3g/l,kh2po
4 5g/l,mgso4·
7h2o 2g/l,feso4·
7h2o 3mg/l,mnso4·
7h2o 4.5mg/l,v
h 3mg/l,v
b1 2mg/l,微量元素混合液2ml/l,消泡剂2滴,其余为水,ph 7.1。
[0131]
发酵培养基成分为:葡萄糖20g/l,木糖20g/l,酵母粉4g/l,(nh4)2so
4 10g/l,kh2po
4 8g/l,mgso4·
7h2o 5g/l,nacl 0.5g/l,feso4·
7h2o 20mg/l,mnso4·
7h2o 4mg/l,cacl2·
2h2o 20mg/l,v
h 1mg/l,v
b1 0.5mg/l,微量元素混合液2ml/l,苯酚红指示剂1%,消
泡剂2滴,其余为水,ph 7.0。
[0132]
微量元素混合液成分为:na2moo4·
2h2o 1g/l,nicl2·
6h2o 1.5g/l,cacl2·
2h2o 8g/l,cuso4·
5h2o 0.5g/l,al2(so4)3·
18h2o 1.5g/l,cocl2·
6h2o 1g/l,znso4·
2h2o 0.5g/l,h3bo
3 0.2g/l,其余为水。
[0133]
摇瓶发酵30h后n
‑
乙酰神经氨酸产量达11.36g/l,生产强度可达0.38g/(l
×
h)。
[0134]
采用发明专利申请202011432150.1:《木糖诱导生产n
‑
乙酰神经氨酸的工程菌及其应用》中构建的菌株e.coli w3110 neu5ac以上述同样的发酵条件进行摇瓶发酵,发酵周期36h,产量达到10.81g/l,生产强度可达0.3g/(l
×
h)。
[0135]
本技术中n
‑
乙酰神经氨酸产量了提升5.09%,生产强度提升26.67%。
[0136]
以上所述实施例仅表达了本发明的几种实施方式,其描述较为具体和详细,但并不能因此而理解为对专利范围的限制。应当指出的是,对于本领域的普通技术人员来说,在不脱离本专利构思的前提下,上述各实施方式还可以做出若干变形、组合和改进,这些都属于本专利的保护范围。因此,本专利的保护范围应以权利要求为准。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1