一种溴芬酸钠新合成方法与流程
:
1.本发明属于药物合成技术领域,更具体地涉及一种溴芬酸钠新合成方法。
背景技术:
2.溴芬酸钠(bromfenac sodium),化学名称为2
‑
氨基
‑3‑
(4
‑
溴苯甲酰基)苯乙酸钠,其结构与酮洛芬和双氯芬酸类似,是最有效的环氧合酶抑制剂之一,能抑制环氧合酶介导的前列腺素类炎症介质的合成,具有较强的消炎镇痛作用,其作用强度是其他非甾体抗炎药的10倍。目前临床上主要作为一种具有消炎作用的滴眼液而使用,用于外眼部及前眼部的炎症性疾病的对症治疗。
[0003][0004]
现已有的溴芬酸钠的合成路线主要有以下几种:
[0005]
专利cn106957237a一种合成溴芬酸钠的方法,其合成路线是以2
‑
氨基
‑4‑
溴二苯甲酮为起始原料经酰化反应、卤乙酰化反应、付克反应后得到目标产物。其合成路线为:
[0006][0007]
此方法用到的溴代乙酸酐、溴乙酰溴使用危险系数较高,挥发出的有机气体对设备厂房腐蚀性极大,另外,工艺中用到氯仿等溶剂,此类溶剂在原料药中残留限度要求高,难以做到。
[0008]
专利cn106397235a,张乐波发明的一种溴芬酸钠的制备方法,其合成以吲哚为起始物料,在二甲亚砜的条件下反应生成3
‑
溴吲哚后水解、氯化、碱解后得到目标产物,其合成路线为:
[0009][0010]
此方法用到的原料吲哚价格较高,收率偏低,产品的酸碱度不稳定。合成过程中用到三氯化硼为高危发烟气体,合成过程中安全风险极大。
[0011]
专利文献ep221753和中国药科大学学报,2003,34(5):405~406)公开了以对溴苯甲腈和吲哚啉为原料,用三氯化硼和三氯化铝为催化剂,进行付克酰化反应,再经氧化、卤化、磷酸水解、氢氧化钠碱解后得到溴芬酸钠,其合成路线为:
[0012][0013]
此方法也是国内生产溴芬酸钠的主流合成路线,但其合成过程中用到二氧化锰,容易导致成品中重金属超标。其合成过程中还用到三氯化硼、n
‑
溴代丁二酰亚胺(nbs)属于高危化学品,其中n
‑
溴代丁二酰亚胺(nbs)具有高毒性,制备过程中容易导致爆炸或人员中毒。
技术实现要素:
[0014]
为解决上述问题,克服现有技术的不足,本发明提供了一种溴芬酸钠新合成方法,能够有效的解决现有技术中因合成方法原因产生的含吲哚环的杂质产生的问题。
[0015]
本发明解决上述技术问题的具体技术方案为:溴芬酸钠新合成方法,其特征在于制备方法包括:
[0016]
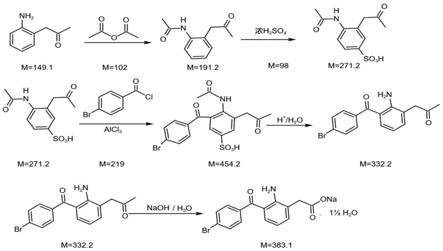
(1)中间体ⅰ的制备:
[0017]
以邻氨基苯乙酸为起始原料,通过酰化反应得到中间体ⅰ;
[0018]
(2)中间体ⅱ的制备;
[0019]
中间体ⅰ经磺化反应得到中间体ⅱ;
[0020]
(3)中间体ⅲ的制备;
[0021]
中间体ⅱ与对溴苯甲酰氯发生取代反应得到中间体ⅲ;
[0022]
(4)溴芬酸的制备;
[0023]
中间体ⅲ经水解后得到溴芬酸;
[0024]
(5)溴芬酸钠的制备;
[0025]
溴芬酸和氢氧化钠反应得到最终的成品溴芬酸钠。
[0026]
所述的中间体ⅰ为邻乙酰氨基苯乙酸;所述中间体ⅱ为2
‑
乙酰氨基
‑
5磺酸基苯乙酸;所述中间体ⅲ为3
‑
(4
‑
溴苯甲酰基)
‑2‑
乙酰氨基
‑5‑
磺酸基苯乙酸。
[0027]
所述的中间体ⅰ的制备:
[0028]
将邻氨基苯乙酸加入溶剂ⅰ内,加入乙酸酐室温反应,缓慢加入反应助剂,生成含有中间体ⅰ混合溶液;中间体ⅰ混合溶液减压浓缩后加入乙醇搅拌均匀,再加入纯化水析晶,抽滤,热风干燥,得到中间体ⅰ;所述溶剂ⅰ为二氯甲烷或甲苯;反应助剂为三乙胺。
[0029]
所述中间体ⅱ的制备:
[0030]
将中间体ⅰ加入溶剂ⅱ中,然后缓慢加入硫酸,反应完毕,加入少量纯化水,静置分层;得到含有中间体ⅱ的混合溶液;含有中间体ⅱ的混合溶液的有机相减压蒸馏后,加入不良溶剂ⅰ,降温析晶,过滤,干燥后得到中间体ⅱ;
[0031]
所述溶剂ⅱ为二氯甲烷或甲苯或甲醇或乙醇的两种溶剂的混合液;所述不良溶剂ⅰ为:甲基叔丁基醚。
[0032]
所述中间体ⅲ的制备:
[0033]
向二氯甲烷中加入中间体ⅱ和三氯化铝,缓慢滴加对溴苯甲酰氯,生成中间体ⅲ的混合溶液,
[0034]
所述溴芬酸的制备;将所述中间体ⅲ的混合溶液减压浓缩至粘稠状,加入盐酸水解,析晶,过滤,纯化水洗涤,热风干燥,得到溴芬酸。
[0035]
将溴芬酸加入溶剂ⅲ中,控温40
‑
50℃,加入氢氧化钠溶液,调节ph值,然后向体系中加入纯化水,充分搅拌,静置、分层;有机层用水萃取一次后,弃去;合并水相,滴加不良溶剂ⅱ析晶,过滤,丙酮洗涤,真空干燥后得到目标产物溴芬酸钠粗品;所述溶剂ⅲ为甲苯或甲醇或乙醇的两种溶剂的混合物,不良溶剂ⅱ为:丙酮或乙二醇二甲醚或异丙醚或甲基叔丁基醚。
[0036]
所述将溴芬酸钠粗品、纯化水加入到反应瓶中,搅拌溶解,加入活性炭脱色,过滤,然后加入不良溶剂ⅱ析晶,抽滤,热风干燥后得到溴芬酸钠成品。所述溴芬酸钠粗品、纯化水的质量比为1:0.8—1.5。
[0037]
本发明的有益效果是:
[0038]
与现有技术比较,本发明所用原辅料采购易得、安全低毒,成本低,此合成路线避免了现有技术中因合成方法原因产生的含吲哚环的杂质产生;
[0039]
本方法制备的溴芬酸钠纯度高,可达99.9%以上,含量在接近100%;
[0040]
本方法避免了现有技术中因使用磷酸或冰醋酸产生酸式盐而导致最终成品ph超标问题,避免了因溴芬酸钠成品制备物料配比原因导致水分过低的质量问题,同时本合成方法简单易控,适于工业化生产。
附图说明:
[0041]
附图1是本发明工艺反应原理化学方程式图;
[0042]
附图2是现有技术工艺生产中产生吲哚杂质ⅰ结构示意图;
[0043]
附图3是现有技术工艺生产中产生吲哚杂质ⅱ结构示意图;附图中:
具体实施方式:
[0044]
在本发明的描述中具体细节仅仅是为了能够充分理解本发明的实施例,但是作为本领域的技术人员应该知道本发明的实施并不限于这些细节。另外,公知的结构和功能没有被详细的描述或者展示,以避免模糊了本发明实施例的要点。对于本领域的普通技术人员而言,可以具体情况理解上述术语在本发明中的具体含义。
[0045]
本发明的具体实施方式:
[0046]
溴芬酸钠新合成方法,其特征在于制备方法包括:
[0047]
(1)中间体ⅰ的制备:
[0048]
以邻氨基苯乙酸为起始原料,通过酰化反应得到中间体ⅰ,即邻乙酰氨基苯乙酸;
[0049][0050]
具体地:
[0051]
将邻氨基苯乙酸加入(二氯甲烷或甲苯)的溶剂ⅰ内,加入乙酸酐室温反应,缓慢加入反应助剂(三乙胺),生成含有中间体ⅰ混合溶液;中间体ⅰ混合溶液减压浓缩后加入乙醇搅拌均匀,再加入纯化水析晶,抽滤,热风干燥,得到中间体ⅰ,即邻乙酰氨基苯乙酸;
[0052]
(2)中间体ⅱ的制备;
[0053]
中间体ⅰ经磺化反应得到中间体ⅱ,即2
‑
乙酰氨基
‑
5磺酸基苯乙酸;
[0054][0055]
具体地:
[0056]
将中间体ⅰ即邻乙酰氨基苯乙酸加入溶剂ⅱ(二氯甲烷或甲苯或甲醇或乙醇的两种溶剂的混合液)中,然后缓慢加入硫酸,反应完毕,加入少量纯化水,静置分层;得到含有中间体ⅱ的混合溶液;将含有中间体ⅱ的混合溶液的有机相减压蒸馏后,加入不良溶剂ⅰ(甲基叔丁基醚),降温析晶,过滤,干燥后得到中间体ⅱ,即2
‑
乙酰氨基
‑
5磺酸基苯乙酸;
[0057]
(3)中间体ⅲ的制备;
[0058]
中间体ⅱ与对溴苯甲酰氯发生取代反应得到中间体ⅲ;
[0059][0060]
具体地:
[0061]
向二氯甲烷中加入中间体ⅱ(2
‑
乙酰氨基
‑
5磺酸基苯乙酸)和三氯化铝,缓慢滴加对溴苯甲酰氯,生成含有中间体ⅲ的混合溶液,即含有3
‑
(4
‑
溴苯甲酰基)
‑2‑
乙酰氨基
‑5‑
磺酸基苯乙酸的的混合溶液,
[0062]
(4)溴芬酸的制备;
[0063]
中间体ⅲ经水解后得到溴芬酸;
[0064][0065]
具体地:
[0066]
将含有所述中间体ⅲ的混合溶液减压浓缩至粘稠状,加入盐酸水解,析晶,过滤,纯化水洗涤,热风干燥,得到溴芬酸。
[0067]
(5)溴芬酸钠的制备;
[0068]
溴芬酸和氢氧化钠反应得到最终的成品溴芬酸钠,
[0069][0070]
具体地:
[0071]
将溴芬酸加入溶剂ⅲ(甲苯或甲醇或乙醇的两种溶剂的混合物)中,控温40
‑
50℃,加入氢氧化钠溶液,调节ph值,然后向体系中加入纯化水,充分搅拌,静置、分层;有机层用水萃取一次后,弃去;合并水相,滴加不良溶剂ⅱ(丙酮或乙二醇二甲醚或异丙醚或甲基叔丁基醚)析晶,过滤,丙酮洗涤,真空干燥后得到目标产物溴芬酸钠粗品;
[0072]
将溴芬酸钠粗品、纯化水加入到反应瓶中,搅拌溶解,加入活性炭脱色,过滤,然后
加入不良溶剂ⅱ析晶,抽滤,热风干燥后得到溴芬酸钠成品。所述溴芬酸钠粗品、纯化水的质量比为1:0.8—1.5。
[0073]
实施例1
[0074]
(1)中间体ⅰ的制备:
[0075]
①
将30g(0.20mol)邻氨基苯乙酸和300ml二氯甲烷加入到反应瓶中,开启搅拌,控温20
‑
30℃开始缓慢滴加乙酸酐40.8g(0.4mol),控制滴加时间在1.5
‑
2h加完;
[0076]
②
滴加乙酸酐30分钟后,从反应瓶的另一加料口滴加三乙胺40.8g(0.40mol);
[0077]
③
滴加完毕,保温反应1小时;
[0078]
④
反应完毕,减压蒸馏,减至内部呈粘稠状,停止减压,加入90g乙醇,搅拌均匀;
[0079]
⑤
将反应液温度控制在20
‑
30℃内,加入720g纯化水,加入时间控制在1
‑
1.5小时内;
[0080]
⑥
保温搅拌析晶1小时,抽滤,滤饼用100ml纯化水洗涤1次,抽滤。滤饼放入热风烘箱,60
‑
70℃干燥10小时,得到邻乙酰氨基苯乙酸(中间体ⅰ)36.6g。
[0081]
(2)中间体ⅱ的制备;
[0082]
①
将35g(0.18mol)中间体ⅰ、二氯甲烷(175g)和乙醇(35g)加入到500ml的四口玻璃反应瓶中,开启搅拌,加热至30
‑
35℃。
[0083]
②
然后向料液中缓慢滴加浓硫酸(70g),控制料液温度不超过40℃。
[0084]
③
保温反应2小时。
[0085]
④
反应完毕,向料液中加入175g纯化水,搅拌5分钟,静置30分钟,分层,水相弃去。
[0086]
⑤
减压浓缩,减压至原体积的50%,停止减压。
[0087]
⑥
控制内温25
‑
30℃向反应瓶中滴加甲基叔丁基醚280g,搅拌析晶,滴加时间在0.5
‑
1小时。降温至5
‑
10℃搅拌析晶1小时,过滤,用150ml甲基叔丁基醚冲洗滤饼,抽干。
[0088]
⑦
50
‑
60℃热风干燥8小时,得到中间体ⅱ即2
‑
乙酰氨基
‑
5磺酸基苯乙酸;44.7g。
[0089]
(3)中间体ⅲ和溴芬酸的制备
[0090]
①
向干燥无水的1000ml反应瓶中加入400ml二氯甲烷、40g(0.14mol)中间体ⅱ、41g三氯化铝,开启搅拌降温至10℃以下。
[0091]
②
向料液中缓慢滴加对溴苯甲酰氯50.7g(0.23mol),控制内温不超过30℃,1.5
‑
2小时加完。
[0092]
③
反应完毕,减压浓缩,将二氯甲烷蒸出,至料液呈粘稠状,生成中间体ⅲ的混合溶液;
[0093]
④
向反应瓶中加入预先配制的10%稀盐酸500ml,控制内温在20
‑
30℃内,1
‑
1.5小时加完。
[0094]
⑤
加完纯化水后继续搅拌析晶1小时,抽滤,滤饼用500ml纯化水洗涤3次,100ml异丙醇冲洗一次,抽干。
[0095]
⑥
设置热风干燥箱温度70℃,将湿品放入干燥箱内干燥12小时,得到溴芬酸44.5g。
[0096]
(4)溴芬酸钠粗品制备
[0097]
①
向1000ml洁净的玻璃反应瓶中加入40g(0.12mol)溴芬酸、加入甲苯250ml和100ml乙醇,搅拌加热至控温30
‑
50℃。
[0098]
②
向料液中加入10%氢氧化钠溶液,调节ph值至11
‑
14。
[0099]
③
然后向料液中加入100ml纯化水,充分搅拌,静置30分钟、分层。
[0100]
④
有机相中加入40ml纯化水,搅拌萃取10分钟后,静置30分钟,分层。
[0101]
⑤
合并水相,有机相弃去。
[0102]
⑥
将水相转移至2000ml反应瓶中,开启搅拌,控温30
‑
40℃滴加丙酮1800ml,2
‑
3小时加完。
[0103]
⑦
缓慢降温至10
‑
15℃,搅拌析晶10小时。
[0104]
⑧
过滤,滤饼用300ml丙酮充分洗涤3次,抽干。
[0105]
⑨
设置热风干燥箱温度在55℃,干燥6小时,得到溴芬酸钠粗品39.3g,纯度99.64%。
[0106]
(5)溴芬酸钠成品制备
[0107]
①
将30g溴芬酸钠粗品、24g纯化水加入到反应瓶中,升温至60℃搅拌溶解,加入活性炭脱色3g,脱色30分钟,过滤。
[0108]
②
滤液降温至15
‑
20℃,然后在2小时以内加入300g丙酮析晶。
[0109]
③
滴加完毕后,将料液降温至0
‑
5℃,搅拌析晶8小时。
[0110]
④
抽滤,滤饼用200ml丙酮充分冲洗2次,抽干。
[0111]
⑤
设置热风干燥箱温度55℃,干燥6小时,得到溴芬酸钠成品27.9g,精制收率93%;纯度:99.92%,含量:100.1%,ph=8.9。
[0112]
实施例2
[0113]
1)中间体ⅰ的制备:
[0114]
①
将15g(0.1mol)邻氨基苯乙酸和150ml二氯甲烷加入到反应瓶中,开启搅拌,控温20
‑
30℃开始缓慢滴加乙酸酐20.4g(0.2mol),控制滴加时间在1.5
‑
2h加完;
[0115]
②
滴加乙酸酐30分钟后,从反应瓶的另一加料口滴加三乙胺20.4g(0.20mol)。
[0116]
③
滴加完毕,保温反应1小时。
[0117]
④
反应完毕,减压蒸馏,减至内部呈粘稠状,停止减压,加入90g乙醇,搅拌均匀。
[0118]
⑤
将反应液温度控制在20
‑
30℃内,加入360g纯化水,加入时间控制在1
‑
1.5小时内。
[0119]
⑥
保温搅拌析晶1小时,抽滤,滤饼用50ml纯化水洗涤1次,抽滤,滤饼放入热风烘箱,60
‑
70℃干燥10小时,得到邻乙酰氨基苯乙酸(中间体ⅰ)17.9g;
[0120]
2)中间体ⅱ的制备
[0121]
①
将15g(0.078mol)中间体ⅰ、二氯甲烷75g、乙醇15g加入到500ml的四口玻璃反应瓶中,开启搅拌,加热至30
‑
35℃。
[0122]
②
然后向料液中缓慢滴加浓硫酸(30g),控制料液温度不超过40℃。
[0123]
③
保温反应2小时。
[0124]
④
反应完毕,向料液中加入75g纯化水,搅拌5分钟,静置30分钟,分层,水相弃去。
[0125]
⑤
减压浓缩,减压至原体积的50%,停止减压。
[0126]
⑥
控制内温25
‑
30℃向反应瓶中滴加甲基叔丁基醚120g,搅拌析晶,滴加时间在0.5
‑
1小时,降温至5
‑
10℃搅拌析晶1小时,过滤,用64ml甲基叔丁基醚冲洗滤饼,抽干。
[0127]
⑦
50
‑
60℃热风干燥8小时,得到中间体ⅱ即2
‑
乙酰氨基
‑
5磺酸基苯乙酸;19.1g;
[0128]
3)中间体ⅲ和溴芬酸的制备
[0129]
①
向干燥无水的500ml反应瓶中加入150ml二氯甲烷、15g(0.052mol)中间体2、15.3g三氯化铝,开启搅拌降温至10℃以下。
[0130]
②
向料液中缓慢滴加对溴苯甲酰氯19g(0.086mol),控制内温不超过30℃,1.5
‑
2小时加完。
[0131]
③
反应完毕,减压浓缩,将二氯甲烷蒸出,至料液呈粘稠状,生成中间体ⅲ的混合溶液;
[0132]
④
向反应瓶中加入10%稀盐酸187ml,控制内温在20
‑
30℃内,1
‑
1.5小时加完。
[0133]
⑤
加完纯化水后继续搅拌析晶1小时。抽滤,滤饼用180ml纯化水洗涤3次,50ml异丙醇充分冲洗一次,抽干。
[0134]
⑥
设置热风干燥箱温度70℃,将湿品放入干燥箱内干燥12小时,得到溴芬酸16.3g;
[0135]
(4)溴芬酸钠粗品制备
[0136]
①
向500ml洁净的玻璃反应瓶中加入15g(0.12mol)溴芬酸、加入甲苯95ml和38ml乙醇,搅拌加热至控温30
‑
50℃。
[0137]
②
向料液中滴加10%氢氧化钠溶液,调节ph值至11
‑
14。
[0138]
③
然后向料液中加入38ml纯化水,充分搅拌,静置30分钟、分层。
[0139]
④
有机相中加入15ml纯化水,搅拌萃取10分钟后,静置30分钟,分层。
[0140]
⑤
合并水相,有机相弃去。
[0141]
⑥
将水相转移至1000ml反应瓶中,开启搅拌,控温30
‑
40℃滴加丙酮680ml,2
‑
3小时加完。
[0142]
⑦
缓慢降温至10
‑
15℃,搅拌析晶10小时。
[0143]
⑧
过滤,滤饼用120ml丙酮充分洗涤3次,抽干。
[0144]
⑨
设置热风干燥箱温度在55℃,干燥6小时,得到溴芬酸钠粗品14.5g,纯度99.71%。
[0145]
(5)溴芬酸钠成品制备
[0146]
①
将10g溴芬酸钠粗品、15g纯化水加入到250ml玻璃反应瓶中,升温至60℃搅拌溶解,加入活性炭脱色1g,脱色30分钟,过滤。
[0147]
②
滤液降温至15
‑
20℃,然后在2小时以内加入100g丙酮析晶。
[0148]
③
滴加完毕后,将料液降温至0
‑
5℃,搅拌析晶8小时。
[0149]
④
抽滤,滤饼用70ml丙酮充分冲洗2次,抽干。
[0150]
⑤
设置热风干燥箱温度55℃,干燥6小时,得到溴芬酸钠成品9.0g,精制收率90%,纯度:99.95%,含量:100%,ph=9.0;
[0151]
实施例3
[0152]
(1)中间体ⅰ的制备:
[0153]
①
将30g(0.20mol)邻氨基苯乙酸和300ml甲苯加入到反应瓶中,开启搅拌,控温20
‑
30℃开始缓慢滴加乙酸酐40.8g(0.4mol),控制滴加时间在1.5
‑
2h加完。
[0154]
②
滴加乙酸酐30分钟后,从反应瓶的另一加料口滴加三乙胺40.8g(0.40mol)。
[0155]
③
滴加完毕,保温反应1小时。
[0156]
④
反应完毕,减压蒸馏,减至内部呈粘稠状,停止减压,加入90g乙醇,搅拌均匀;
[0157]
⑤
将反应液温度控制在20
‑
30℃内,加入720g纯化水,加入时间控制在1
‑
1.5小时内;
[0158]
⑥
保温搅拌析晶1小时,抽滤,滤饼用100ml纯化水洗涤1次,抽滤。滤饼放入热风烘箱,60
‑
70℃干燥10小时,得到邻乙酰氨基苯乙酸(中间体ⅰ)34.5g;
[0159]
(2)中间体ⅱ的制备
[0160]
①
将30g(0.11mol)中间体ⅰ、二氯甲烷(150g)和乙醇(30g)加入到500ml的四口玻璃反应瓶中,开启搅拌,加热至30
‑
35℃。
[0161]
②
然后向料液中缓慢滴加浓硫酸(60g),控制料液温度不超过40℃。
[0162]
③
保温反应2小时。
[0163]
④
反应完毕,向料液中加入150g纯化水,搅拌5分钟,静置30分钟,分层,水相弃去。
[0164]
⑤
减压浓缩,减压至原体积的50%,停止减压。
[0165]
⑥
控制内温25
‑
30℃向反应瓶中滴加甲基叔丁基醚240g,搅拌析晶,滴加时间在0.5
‑
1小时,降温至5
‑
10℃搅拌析晶1小时,过滤,用130ml甲基叔丁基醚冲洗滤饼,抽干。
[0166]
⑦
50
‑
60℃热风干燥8小时,得到中间体ⅱ即2
‑
乙酰氨基
‑
5磺酸基苯乙酸;38.7g;
[0167]
(3)中间体ⅲ和溴芬酸的制备
[0168]
①
向干燥无水的1000ml反应瓶中加入350ml甲苯、35g(0.14mol)中间体2、35.8g三氯化铝,开启搅拌降温至10℃以下。
[0169]
②
向料液中缓慢滴加对溴苯甲酰氯44.3g(0.20mol),控制内温不超过30℃,1.5
‑
2小时加完。
[0170]
③
反应完毕,减压浓缩,将二氯甲烷蒸出,至料液呈粘稠状,生成中间体ⅲ的混合溶液;
[0171]
④
向反应瓶中加入预先配制的10%稀盐酸440ml,控制内温在20
‑
30℃内,1
‑
1.5小时加完。
[0172]
⑤
加完纯化水后继续搅拌析晶1小时,抽滤,滤饼用440ml纯化水洗涤3次,100ml异丙醇冲洗一次,抽干。
[0173]
⑥
设置热风干燥箱温度70℃,将湿品放入干燥箱内干燥12小时,得到溴芬酸36g;
[0174]
(4)溴芬酸钠粗品制备
[0175]
①
向1000ml洁净的玻璃反应瓶中加入35g(0.105mol)溴芬酸、加入甲苯220ml和87ml乙醇,搅拌加热至控温30
‑
50℃。
[0176]
②
向料液中加入10%氢氧化钠溶液,调节ph值至11
‑
14。
[0177]
③
然后向料液中加入87ml纯化水,充分搅拌,静置30分钟、分层。
[0178]
④
有机相中加入35ml纯化水,搅拌萃取10分钟后,静置30分钟,分层。
[0179]
⑤
合并水相,有机相弃去。
[0180]
⑥
将水相转移至2000ml反应瓶中,开启搅拌,控温30
‑
40℃滴加乙二醇二甲醚1580ml,2
‑
3小时加完。
[0181]
⑦
缓慢降温至10
‑
15℃,搅拌析晶10小时。
[0182]
⑧
过滤,滤饼用260ml乙二醇二甲醚充分洗涤3次,抽干。
[0183]
⑨
设置热风干燥箱温度在55℃,干燥6小时,得到溴芬酸钠粗品34.9g,纯度
99.58%。
[0184]
(5)溴芬酸钠成品制备
[0185]
①
将30g溴芬酸钠粗品、30g纯化水加入到反应瓶中,升温至60℃搅拌溶解,加入活性炭脱色3g,脱色30分钟,过滤。
[0186]
②
滤液降温至15
‑
20℃,然后在2小时以内加入300g乙二醇二甲醚析晶。
[0187]
③
滴加完毕后,将料液降温至0
‑
5℃,搅拌析晶8小时。
[0188]
④
抽滤,滤饼用200ml乙二醇二甲醚充分冲洗2次,抽干。
[0189]
⑤
设置热风干燥箱温度55℃,干燥6小时,得到溴芬酸钠成品28.5g,精制收率95%。纯度:99.93%,含量:100%。ph=8.7。
[0190]
为了更加直观的展现本发明的工艺优势,特以本发明一种溴芬酸钠新合成方法与现有技术ⅰ利用(按照国家发明专利cn106397235a,以吲哚为起始物料,在二甲亚砜的条件下反应生成3
‑
溴吲哚后水解、氯化、碱解后得到目标产物,以援引方式引入),
[0191]
现有技术ⅱ利用(按照发明专利ep0221753,以对溴苯甲腈和吲哚啉为原料,用三氯化硼和三氯化铝催化下进行付克酰化反应,再经氧化、卤化、磷酸水解、氢氧化钠碱解后得到溴芬酸钠,以援引方式引入),
[0192]
表1:不同工艺产生吲哚杂质的对比
[0193][0194][0195]
试验结果如下:
[0196]
现有技术ⅰ和现有技术ⅱ由于工艺本身的原理存在的缺陷,导致了生成溴芬酸钠的过程中产生了吲哚杂质,值得注意的是吲哚杂质与中间体物化性质相似不易分离,进而影响了产品的纯度;
[0197]
为了更加直观的展现本发明的工艺优势,特以本发明采用一种溴芬酸钠新合成方法和相同工艺采用等效替换的方法进行对比,
[0198]
对比例一:
[0199]
制备方法同现有技术ⅰ,所不同的是:本对比例的制备过程中,酸水解采的磷酸替换成盐酸;
[0200]
对比例二:
[0201]
制备方法同现有技术ⅱ,所不同的是:本对比例的制备过程中,酸水解采的磷酸替换成盐酸;
[0202]
对比例三:
[0203]
制备方法同本发明,所不同的是:本对比例的制备过程中,酸水解采的盐酸替换成磷酸;
[0204]
溴芬酸钠成品的碱度检测标准ph=8.5
‑
9.5;
[0205]
取本品1.0g,加新沸过的冷水20ml溶解后,照(《药品检验操作规程》(例通则)通则
17)ph值测定法迅速测定。
[0206]
溴芬酸钠有关物质(纯度)的检测方法:
[0207]
色谱条件及系统适用性试验:
[0208]
用十八烷基硅烷键合硅胶为填充剂(5μm,4.6
×
250mm),以磷酸盐缓冲液(取磷酸氢二铵2.5g,加水溶解并稀释至1000ml,用磷酸调节ph值至7
‑
8)
‑
乙腈(75:20)为流动相a,磷酸盐缓冲液(取磷酸氢二铵2.5g,加水溶解并稀释至1000ml,用磷酸调节ph值至7.3)
‑
乙腈(20:70)为流动相b,按下表进行梯度洗脱,调整流速使溴芬酸钠峰的保留时间约为18分钟,检测波长为266nm。
[0209]
表2:不同工艺及中间体酸水解工艺碱度检测指标对照数据
[0210][0211]
试验结果如下:
[0212]
采用现有的生产溴芬酸钠的工艺,由于工艺的限定,对于预酸化的中间产物3
‑
溴吲哚或3
‑
溴
‑7‑
(4
‑
溴苯甲酰基)吲哚进行酸化时;采用的磷酸产生酸式盐,会导致ph超标,不满足溴芬酸钠成品的碱度检测标准ph=8.5
‑
9.5;
[0213]
而对比例一和对比例二的数据分析可知,采用现有的生产溴芬酸钠的工艺,并将酸化的磷酸替换成本发明的盐酸,导致了发生副反应,得不到目标产物;
[0214]
对比例三的数据分析可知,采用中间体3
‑
(4
‑
溴苯甲酰基)
‑2‑
乙酰氨基
‑5‑
磺酸基苯乙酸进行酸化,替换成磷酸,ph≥10.1,不满足溴芬酸钠成品的碱度检测标准ph=8.5
‑
9.5。
[0215]
为了更加直观的展现本发明的精制工艺优势,特以本发明采用一种溴芬酸钠新合成方法和相同工艺采用等效替换的方法进行对比,
[0216]
实施例四:
[0217]
精制方法同本发明实施例三,所不同的是:本对比例的制备过程中,精制过程中将30g溴芬酸钠粗品、30g纯化水替换成:30g溴芬酸钠粗品、24g纯化水;
[0218]
实施例五:
[0219]
精制方法同本发明实施例三,所不同的是:本对比例的制备过程中,精制过程中将30g溴芬酸钠粗品、30g纯化水替换成:30g溴芬酸钠粗品、45g纯化水;
[0220]
对比例四:
[0221]
精制方法同本发明实施例三,所不同的是:本对比例的制备过程中,精制过程中将30g溴芬酸钠粗品、30g纯化水替换成;30g溴芬酸钠粗品、15g纯化水;
[0222]
对比例五:
[0223]
精制方法同本发明实施例三,所不同的是:本对比例的制备过程中,精制过程中将30g溴芬酸钠粗品、30g纯化水替换成;30g溴芬酸钠粗品、60g纯化水;
[0224]
表3:不同精制工艺的溴芬酸钠检测指标对照数据
[0225] 溴芬酸钠粗品纯化水精制总收率含水量实施例三303095%7.3%实施例四302496%6.8%实施例五304593.3%7.5%对比例四301591%6.0%对比例五306072%8.3%
[0226]
试验结果如下:
[0227]
由上述实验结果分析可知:
[0228]
从实施例三
‑
实施例五可知:溴芬酸钠粗品用量:纯化水用量=1:0.8—1.5范围内,既能获得优越的总收率,又可以保证溴芬酸钠的合适含水量;
[0229]
从对比例四
‑
实施例五可知:溴芬酸钠粗品用量与纯化水用量的比例,随着纯化水用量的增加,过多的水分会导致溴芬酸钠的流失,进而会导致溴芬酸钠的总收率较低;
[0230]
溴芬酸钠粗品用量与纯化水用量的比例,随着纯化水用量的减少,会导致溴芬酸钠的成品含水量较低,进而不满足溴芬酸钠有关物质的检测指标。
[0231]
综上所述:
[0232]
本发明所用原辅料采购易得、安全低毒,成本低,此合成路线避免了现有技术中因合成方法原因产生的含吲哚环的杂质产生;
[0233]
制备的溴芬酸钠纯度高,可达99.9%以上,含量在接近100%;
[0234]
创造性的采用全新的工艺,制备了中间体3
‑
(4
‑
溴苯甲酰基)
‑2‑
乙酰氨基
‑5‑
磺酸基苯乙酸,并将该中间体采用盐酸酸化,避免了现有技术中因使用磷酸或冰醋酸产生酸式盐而导致最终成品ph超标问题;
[0235]
通过调整溴芬酸钠粗品与纯化水的用量避免了因溴芬酸钠成品制备物料配比原因导致水分过低的质量问题,同时本合成方法简单易控,适于工业化生产。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1