一种高纯度环丙基甲基酮的制备方法与流程
1.本发明涉及医药中间体领域,具体涉及一种环丙基甲基酮的制备方法。
背景技术:
2.环丙基甲基酮(cpmo)是合成环丙氟哌酸类药物的重要中间体,也是合成抗艾滋特效药依法韦仑、绿色农药嘧菌环胺以及植物生长调节剂环唑醇的重要中间体。
[0003][0004]
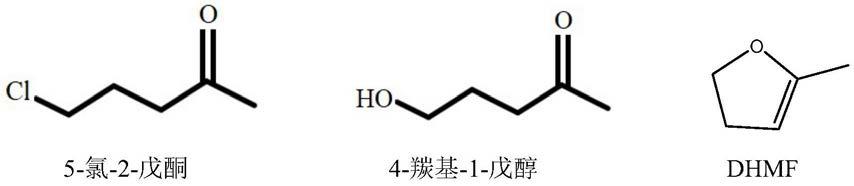
环丙基甲基酮的合成通常以5
‑
氯
‑2‑
戊酮为原料经环合得到粗品,然后精馏分离得到高纯度产品。例如,专利cn200610098154公开了一种环丙基甲基酮的制备方法,是将乙酰正丙醇加到盐酸溶液中,发生氯化反应,采用分离耦合技术,将5-氯-2-戊酮蒸出;5-氯-2-戊酮和碱发生环合反应,生成环丙基甲基酮。又如专利申请cn201210099170同样公开了一种环丙基甲基酮的制备方法,其采用了2
‑
乙酰基
‑
γ
‑
丁内酯为原料和盐酸进行氯化反应,制得5
‑
氯
‑2‑
戊酮,5
‑
氯
‑2‑
戊酮再和液碱进行环合反应,得环丙基甲基酮粗品,再经精馏得环丙基甲基酮产品。而专利cn201310135830涉及一种环丙乙炔重要中间体环丙基甲基酮的制备方法。具体以5
‑
氯
‑2‑
戊酮为起始原料,在碱性条件下,在多段反应器中,采用反应精馏技术来进行反应,反应温度控制为90
‑
150℃,在多段反应器中收集得到环丙基甲基酮来实现。专利申请cn201911235730涉及一种环丙基甲基酮的合成方法,包括以下步骤:1)将2
‑
甲基呋喃在加氢催化剂、氢气和水存在下采用一锅法进行氢化水解,制得乙酰正丙醇;2)将步骤1)所制得的乙酰正丙醇进行盐酸氯化反应,制得5
‑
氯
‑2‑
戊酮;3)将步骤2)所制得的5
‑
氯
‑2‑
戊酮的粗品在碱性条件下进行关环反应,即得环丙基甲基酮。
[0005]
上述以5
‑
氯
‑2‑
戊酮为原料经环合得到的环丙基甲基酮粗品中,除了未反应完全的5
‑
氯
‑2‑
戊酮或4
‑
羰基
‑1‑
戊醇,常伴有大量低沸点有机萃取剂、废酸水、废碱水、固废残渣及dhmf等各类副产物。而其中杂质dhmf的分离是关键。
[0006][0007]
专利de6002046提供了一种从环丙基甲基酮反应产物中分离dhmf的方法,即向含环丙基甲基酮、dhmf及酸催化剂的反应物中加入醇,水或水/醇混合物,并使dhmf与其在10~80℃下反应,经精馏纯化后,有效获得gc纯度>99%的环丙基甲基酮。此发明将反应体系中的dhmf转化为4
‑
羰基
‑1‑
戊醇,可以一定程度减少dhmf,同时反应体系引入了水和醇等杂质,需要考虑新杂质和水对分离的影响。
[0008]
专利us6045662指出4
‑
羰基
‑1‑
戊醇热不稳定,受热后可转化为dhmf,而dhmf在和水存在且加热条件下可以转化为4
‑
羰基
‑1‑
戊醇,两者可逆。该专利通过控制釜温低于140℃(目的是减少dhmf的生成),并采用高分离效率的精馏塔连续精馏得到纯度大于99.0%的环丙基甲基酮。其中,式i为dhmf和4
‑
羰基
‑1‑
戊醇相互转换的方程式。
[0009][0010]
综上,上述方法只在一定程度上减缓或者减少了dhmf的生成,因体系中存在的4
‑
羰基
‑1‑
戊醇,随着精馏进程,持续不断地生成dhmf,导致常规条件下难以获得更高纯度(gc含量>99.9%)的环丙基甲基酮。而要获得更高纯度的产品需要反复精馏,如此势必会影响产品收率,浪费能源。
[0011]
此外,专利申请201910859953.6提供一种加盐萃取及精馏提纯环丙基甲基酮的装置及方法,目的在于提供一种节能的环丙基甲基酮的分离提纯工艺,提纯方法采取萃取与精馏技术耦合,其中,萃取采用两步萃取工艺,先将一种萃取剂按比例加入原料中,搅拌
‑
静置
‑
分层,排出水层,再次向有机层按比例加入另一种萃取剂,搅拌
‑
静置
‑
分层,排出水层,将有机层泵入超重力精馏装置进行精馏,塔底得环丙基甲基酮产品。缩短了分离时间,降低了环丙基甲基酮分离提纯成本。该方法采取精馏和萃取结合的方法提纯环丙基甲基酮,相应的装置和方法都变得高度复杂化。
[0012]
因此,本领域需要一种新的高纯度环丙基甲基酮的制备方法,尤其是一种能够通过精馏高效率地将dhmf与环丙基甲基酮分开,从而得到高纯度环丙基甲基酮的方法。
技术实现要素:
[0013]
因此,本发明提供一种高纯度环丙基甲基酮的制备方法,所述方法包括先由5
‑
氯
‑2‑
戊酮经过化学反应制备得到环丙基甲基酮反应液,再在所述环丙基甲基酮反应液中加入自由基引发剂,使得环丙基甲基酮反应液中的4,5
‑
二氢
‑2‑
甲基呋喃(dhmf)聚合反应形成2
‑
甲基呋喃聚合物,因而所述环丙基甲基酮反应液变成dhmf低含量混合液,再将所述dhmf低含量混合液经精馏分离得到高纯度环丙基甲基酮产品。
[0014]
在一种具体的实施方式中,所述高纯度环丙基甲基酮产品的gc含量≥99.9%。
[0015]
在一种具体的实施方式中,所述环丙基甲基酮反应液中含有环丙基甲基酮、4,5二氢
‑2‑
甲基呋喃、5
‑
氯
‑2‑
戊酮和4
‑
羰基
‑1‑
戊醇,且自由基引发剂的用量为环丙基甲基酮反应液总质量的0.0001~2wt%,优选0.001~1wt%,更优选0.02~0.5wt%。
[0016]
在一种具体的实施方式中,所述自由基引发剂为过氧化二苯甲酰、偶氮二异丁腈、偶氮二异戊腈、偶氮二异庚腈、偶氮二异丁酸二甲酯、过氧化二异丙苯和二叔丁基过氧化物中的一种或多种。
[0017]
在一种具体的实施方式中,所述聚合反应的温度为10~100℃,优选40~80℃。
[0018]
在一种具体的实施方式中,所述聚合反应的时间为1小时以上,优选5小时以上,更
优选12~24小时。
[0019]
在一种具体的实施方式中,所述精馏分离的釜底温度或塔底温度为120~180℃,精馏的压力为常压。
[0020]
本发明通过对环丙基甲基酮粗品简单处理后,精馏得到更高纯度的环丙基甲基酮,提高精馏分离效率。
[0021]
本发明将环丙基甲基酮粗品中的dhmf在自由基引发剂的作用下转化成2
‑
甲基呋喃聚合物(反应方程式见式ii),彻底解决dhmf杂质的分离问题。
[0022][0023]
与现有技术相比,本项目的有益效果至少包括:
[0024]
1)本发明在环丙基甲基酮反应液(cpmo粗品)中加入自由基引发剂,将杂质dhmf转化为聚合物,彻底解决dhmf难分离的问题。
[0025]
2)本发明中加入的自由基引发剂会随着温度升高而被降解,不会对cpmo的精馏产生任何负面影响。
[0026]
3)本发明中产品cmpo的精馏收率大于90%,产品gc纯度大于99.9%,该方法在提高产品精馏效率的同时显著降低能耗。
具体实施方式
[0027]
实施例1
[0028]
环丙基甲基酮反应液1000g,过氧化二苯甲酰0.5g,搅拌升温至40℃反应12h,然后转移至精馏釜中,加热升温,当气相温度为80
‑
90℃,回流分水,待无水分出后,采出前馏份,气相温度为114℃,回流比4:1
‑
2:1,取样检测,气相含量≥99.9%,回流比1:1,接产品,无馏份蒸出,停止精馏,得产品924g。
[0029]
实施例2
[0030]
环丙基甲基酮反应液1000g,偶氮二异丁腈1.0g,搅拌升温至60℃反应8h,然后转移至精馏釜中,加热升温,当气相温度为80
‑
90℃,回流分水,待无水分出后,采出前馏份,气相温度为114℃,回流比4:1
‑
2:1,取样检测,气相含量≥99.9%,回流比1:1,接产品,无馏份蒸出,停止精馏,得产品947g。
[0031]
实施例3
[0032]
环丙基甲基酮反应液1000g,过氧化二苯甲酰0.5g,搅拌升温至40℃反应12h,然后转移至精馏釜中,加热升温,当气相温度为80
‑
90℃,回流分水,待无水分出后,采出前馏份,气相温度为114℃,回流比4:1
‑
2:1,取样检测,气相含量≥99.9%,回流比1:1,接产品,无馏份蒸出,停止精馏,得产品915g。
[0033]
实施例4
[0034]
环丙基甲基酮反应液1000g,过氧化二异丙苯0.75g,搅拌升温至80℃反应12h,然
后转移至精馏釜中,加热升温,当气相温度为80
‑
90℃,回流分水,待无水分出后,采出前馏份,气相温度为114℃,回流比4:1
‑
2:1,取样检测,气相含量≥99.9%,回流比1:1,接产品,无馏份蒸出,停止精馏,得产品903g。
[0035]
对比例1
[0036]
环丙基甲基酮反应液1000g,加入精馏釜中,加热升温,当气相温度为80
‑
90℃,回流分水,待无水分出后,采出前馏份,气相温度为114℃,回流比4:1
‑
2:1,取样检测,气相含量≥99.9%,回流比1:1,接产品,无馏份蒸出,停止精馏,得产品650g,含量99.9%。一次精馏收率仅65%。
[0037]
表1为实施例1~4和对比例1的结果对比表格。
[0038]
表1
[0039][0040]
从表1的反应结果可见,实施例的前馏分中杂质dhmf含量均为0.16%以内,而对比例1中的前馏分中杂质dhmf含量为1.13%,相应的,实施例中的前馏分质量的占比均在4.2%以内,而对比例1中的前馏分质量的占比达到30%。再看一次性精馏的cpmo产品的收率及其cpmo的gc含量,可见实施例1~4中在确保产品中cpmo的gc含量高于99.9%的前提下,一次性精馏的cpmo产品的收率均达到90%以上,而相应gc纯度的对比例1中一次性精馏的cpmo产品的收率仅65%。由此可见,本发明在环丙基甲基酮反应液中加入自由基引发剂,将杂质dhmf转化为聚合物,彻底解决了杂质dhmf与产品cpmo难分离的问题,且本发明中加入的自由基引发剂会随着温度升高而降解,其加入不会对cpmo的精馏产生任何负面影响。本发明中产品cmpo的一次性精馏收率大于90%,产品cmpo的gc纯度大于99.9%,该方法在提高产品精馏效率的同时显著降低能耗。因而,本发明提供了一种全新且高效率的高纯度环丙基甲基酮的制备方法。
[0041]
以上所述实施例仅为本发明的优选实施方式,但本发明的保护范围并不局限于此,任何熟悉本领域的技术人员在本发明揭示的技术范围内,根据本发明的技术方案及发明构思进行的等同替换或修改,都应涵盖在本发明的保护范围之内。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1