细菌生物被膜核心胞外多糖裂解酶PelAN及其制备方法和应用
细菌生物被膜核心胞外多糖裂解酶pelan及其制备方法和应用
技术领域
1.本发明属于微生物工程技术领域,具体涉及细菌生物被膜核心胞外多糖裂解酶pelan及其制备方法和应用。
背景技术:
2.大多数革兰氏阴性菌可与其分泌物如胞外多糖、胞外蛋白和胞外dna等,形成结构坚固的聚集体,即生物被膜。生物被膜被认为是导致抗生素耐药性的重要因素之一,可广泛存在于水源、土壤以及各类食品之中,导致严重的公共卫生问题。
3.pel是一种由n
‑
乙酰
‑
d
‑
氨基葡萄糖和n
‑
乙酰基
‑
d
‑
半乳糖胺组成的阳离子胞外多糖,是假单胞菌生物被膜形成必不可少的成分,与该菌生物被膜厚度、粘附性和细胞聚集性密切相关,由pel系统转运合成。该pel系统中的伴侣蛋白pela含有1个糖苷水解酶结构域pelan,其重组蛋白(pelan)能够从根本上瓦解pel多糖,抑制和清除革兰氏阴性菌生物被膜。现阶段关于pel胞外多糖抑制剂的研究还相对较少,尤其是在食品科学领域的相关报道。
技术实现要素:
4.基于pelan的生物学特性及为弥补其工业化制备的技术空白,本发明的主要目的是提供一种细菌生物被膜核心胞外多糖裂解酶pelan的制备方法,该方法步骤简单,操作方便,适用于大量提取高纯度、高活性的pelan蛋白。
5.本发明的另一目的是提供上述方法得到的细菌生物被膜核心胞外多糖裂解酶pelan在抑制细菌生物被膜形成或靶向清除细菌生物被膜中的应用。
6.为实现上述目的,本发明通过以下技术方案实现:
7.本发明提供一种细菌生物被膜核心胞外多糖裂解酶pelan的制备方法,包含以下步骤:
8.(1)获取细菌的全基因组序列并设计编码细菌生物被膜核心胞外多糖裂解酶pelan的基因序列,将其克隆到pet28b表达载体中,得到pet28b
‑
pelan重组表达载体;
9.(2)对步骤(1)中所述pet28b
‑
pelan重组表达载体测序确认后,导入到e.coli bl21(de3)感受态细胞中,得到de3工程菌株;
10.(3)将步骤(2)中所述de3工程菌株扩大培养,加入iptg(异丙基
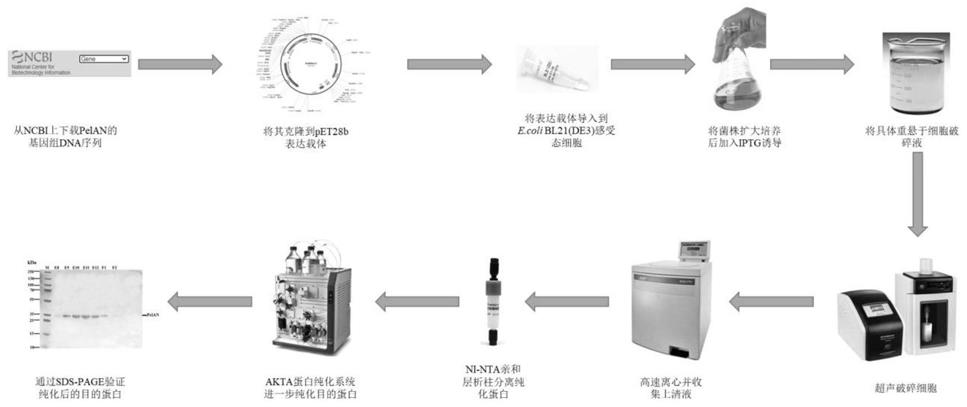
‑
β
‑
d
‑
硫代半乳糖苷)诱导表达,离心收集菌体;
11.(4)将步骤(3)中收集的菌体重悬于细胞破碎液中,冰浴下超声破碎;
12.(5)将步骤(4)中破碎液高速离心,收集上清液;
13.(6)将步骤(5)中上清液依次经ni
‑
nta亲和层析柱分离纯化和akta蛋白纯化系统纯化,收集pelan蛋白,通过sds
‑
page验证纯化后得到。
14.优选地,所述细菌为革兰氏阴性细菌,选自铜绿假单胞菌pa14、荧光假单胞菌
nbrc15842或恶臭假单胞菌jbc17。
15.优选地,步骤(1)中,编码细菌生物被膜核心胞外多糖裂解酶pelan的基因序列如seq id no:2、4或6所示,由ncbi数据库获取所述细菌的全基因组序列后通过phyre2同源建模分析确定pelan氨基酸位点和稀有密码子偏好性优化得到。
16.优选地,步骤(2)中,还包括所述de3工程菌株经过终浓度为50μg/ml的卡那霉素进行阳性筛选的步骤。
17.优选地,步骤(3)中,所述de3工程菌株扩大培养至菌液浓度od
600
为0.55
–
0.6。
18.优选地,步骤(3)中,iptg诱导表达至菌体的终浓度为1mmol/l,诱导条件为20℃过夜诱导培养18h。
19.优选地,步骤(4)中,所述细胞破碎液的组分为20mm tris、300mm nacl、10mm imidazole、5%glycerol、30mg/ml溶菌酶、一片罗氏蛋白酶抑制剂和一勺dna酶。
20.优选地,步骤(6)中,所述ni
‑
nta亲和层析柱含有缓冲液1、缓冲液2和缓冲液3;其中,所述缓冲液1的组分为20mm tris、300mm nacl、10mm imidazole和5%glycerol;所述缓冲液2的组分为20mm tris、500mm nacl、35mm imidazole和5%glycerol;所述缓冲液3的组分为20mm tris、500mm nacl、300mm imidazole和5%glycerol。
21.优选地,步骤(6)中,所述akta蛋白纯化系统含有缓冲液4,其组分为25mm tris、200mm nacl和5%glycerol。
22.本发明还提供一种细菌生物被膜核心胞外多糖裂解酶pelan,通过上述任一所述细菌生物被膜核心胞外多糖裂解酶pelan的制备方法得到。
23.优选地,所述细菌生物被膜核心胞外多糖裂解酶pelan选自铜绿假单胞菌pa14生物被膜核心胞外多糖裂解酶pelan
‑
pa、荧光假单胞菌nbrc15842生物被膜核心胞外多糖裂解酶pelan
‑
pf或者恶臭假单胞菌jbc17生物被膜核心胞外多糖裂解酶pelan
‑
pp。
24.本发明还提供上述任一所述细菌生物被膜核心胞外多糖裂解酶pelan在抑制细菌生物被膜形成或靶向清除细菌生物被膜中的应用。
25.优选地,所述细菌为革兰氏阴性细菌,选自铜绿假单胞菌pa14、荧光假单胞菌nbrc15842或恶臭假单胞菌jbc17。
26.优选地,所述细菌生物被膜核心胞外多糖裂解酶pelan为细菌生物被膜清除剂或细菌生物被膜消毒剂。
27.与现有技术相比,本发明的有益效果在于:
28.(1)本发明基于pelan的生物学特性构建一种细菌生物被膜核心胞外多糖裂解酶pelan的制备方法,步骤简单且操作方便,适用于大量提取高纯度、高活性的pelan蛋白,弥补其工业化制备的技术空白。
29.(2)本发明制备的细菌生物被膜核心胞外多糖裂解酶pelan可用于抑制细菌生物被膜形成或靶向清除细菌生物被膜,为pelan靶向清除革兰氏阴性细菌生物被膜奠定基础,用于食品等工业中控制生物被膜的风险及其危害。
30.以下将结合附图对本发明的构思及产生的技术效果进一步说明,以充分地了解本发明的目的、特征和效果。
附图说明
31.图1为本发明中细菌生物被膜核心胞外多糖裂解酶pelan的制备流程示意图。
32.图2为本发明中pet28b
‑
pelan表达载体的构建示意图。
33.图3为实施例1中铜绿假单胞菌pa14裂解酶pelan
‑
pa的凝胶过滤层析(a)和sds
‑
page分析(b)。
34.图4为实施例2中荧光假单胞菌nbrc15842裂解酶pelan
‑
pf的凝胶过滤层析(a)和sds
‑
page分析(b)。
35.图5为实施例3中恶臭假单胞菌jbc17裂解酶pelan
‑
pp的凝胶过滤层析(a)和sds
‑
page分析(b)。
36.图6为实施例4中胞外多糖裂解酶pelan
‑
pa、pelan
‑
pf和pelan
‑
pp抑制铜绿假单胞菌pa14生物被膜的测试图。
具体实施方式
37.下面通过具体实例和附图,对本发明的技术方案做进一步的菌体说明。应当理解,以下描述的具体实例仅用于解释文本发明,并不用于限定本发明。所描述的实施例仅是本发明一部分实施例,而不是全部的实施例,基于本发明中的实施例,本领域的普通技术人员咋没有做出创造性劳动前提下所获得的所有其他实施例,都属于本发明保护的范围。
38.如没有特别说明,本发明采用的试剂、方法和设备均为本技术领域常规试剂、方法和设备。所用试剂均为分析纯,水为去离子水。
39.如图1和2所示,描述了本发明中细菌生物被膜核心胞外多糖裂解酶pelan的制备方法,包含以下步骤:
40.(1)获取细菌的全基因组序列并设计编码细菌生物被膜核心胞外多糖裂解酶pelan的基因序列,将其克隆到pet28b表达载体中,得到pet28b
‑
pelan重组表达载体;
41.(2)对步骤(1)中pet28b
‑
pelan重组表达载体测序确认后,导入到e.coli bl21(de3)感受态细胞中,得到de3工程菌株;
42.(3)将步骤(2)中de3工程菌株扩大培养,加入iptg(异丙基
‑
β
‑
d
‑
硫代半乳糖苷)诱导表达,离心收集菌体;
43.(4)将步骤(3)中收集的菌体重悬于细胞破碎液中,冰浴下超声破碎;
44.(5)将步骤(4)中破碎液高速离心,收集上清液;
45.(6)将步骤(5)中上清液依次经ni
‑
nta亲和层析柱分离纯化和akta蛋白纯化系统纯化,收集pelan蛋白,通过sds
‑
page验证纯化后得到。
46.一些实施例中,细菌为革兰氏阴性细菌,选自铜绿假单胞菌pa14、荧光假单胞菌nbrc15842或恶臭假单胞菌jbc17。
47.一些实施例中,步骤(1)中,编码细菌生物被膜核心胞外多糖裂解酶pelan的基因序列由ncbi数据库获取细菌的全基因组序列后通过phyre2同源建模分析确定pelan氨基酸位点和稀有密码子偏好性优化得到。
48.一些实施例中,步骤(2)中,还包括de3工程菌株经过终浓度为50μg/ml的卡那霉素进行阳性筛选的步骤。
49.一些实施例中,步骤(3)中,de3工程菌株扩大培养至菌液浓度od
600
为0.55
–
0.6;
iptg诱导表达至菌体的终浓度为1mmol/l,诱导条件为20℃过夜诱导培养18h。
50.一些实施例中,步骤(4)中,细胞破碎液的组分为20mm tris、300mm nacl、10mm imidazole、5%glycerol、30mg/ml溶菌酶、一片罗氏蛋白酶抑制剂和一勺dna酶。
51.一些实施例中,步骤(6)中,ni
‑
nta亲和层析柱含有缓冲液1、缓冲液2和缓冲液3;其中,缓冲液1的组分为20mm tris、300mm nacl、10mm imidazole和5%glycerol;缓冲液2的组分为20mm tris、500mm nacl、35mm imidazole和5%glycerol;缓冲液3的组分为20mm tris、500mm nacl、300mm imidazole和5%glycerol;akta蛋白纯化系统含有缓冲液4,其组分为25mm tris、200mm nacl和5%glycerol。
52.实施例1
53.一种铜绿假单胞菌pa14生物被膜核心胞外多糖裂解酶pelan
‑
pa的制备方法,包含以下步骤:
54.1)获取并设计细菌pelan
‑
pa基因组dna:从ncbi数据库下载获得铜绿假单胞菌pa14的全基因组序列(protein_id=eot14801.1),并通过phyre2对其进行同源建模分析,发现其糖苷水解酶域位于第47
–
303位氨基酸(seq id no:1:ggpssvafwyaerpplaelsqfdwvvleaahlkpadvgylkeqgstpfaylsvgefdgdaaaiadsglargksavrnqawnsqvmdlaapswrahllkraaelrkqgyaglfldtldsfqlqaeerregqrralasflaqlhrqepglklffnrgfevlpelpgvasavavesihagwdaaagqyrevpqddrdwlkghldalraqgmpivaidylpperrdearalaarlrsegyvpfvstpaldylgvsdvevqp),参照大肠杆菌遗传密码子频率表,通过在线密码子优化软件e.coli codon usage analyzer 2.1对序列进行稀有密码子偏好性优化,得到编码铜绿假单胞菌pa14生物被膜核心胞外多糖裂解酶pelan
‑
pa的核苷酸序列(seq id no:2:ggcgggccgtccagcgtggcgttctggtacgccgagcggccgccgctggccgagctttcccagttcgactgggtggtgctcgaagcggcgcacctcaagccggccgatgtcgggtatctgaaagagcagggcagcacgcccttcgcctatctgtcggtcggcgagttcgacggcgacgccgccgccatcgccgatagcggcctggcccggggcaagagcgcggtccgcaaccaagcctggaacagccaggtaatggacctcgccgcgccgagttggcgggcgcacctgctcaagcgcgccgcggagctgcgcaaacagggctacgccggcctgttcctcgataccctggacagcttccagctacaggccgaggagcgccgcgagggccagcgccgggcgctggccagtttcctcgcccagctgcatcgccaggagccgggcctcaagctgtttttcaatcgcggtttcgaagtgctgccggagttgcccggcgtcgcgtcggcggtggccgtggagtcgatccatgccggttgggacgccgctgccgggcaataccgcgaggtgccccaggacgatcgcgattggctgaagggtcacctggatgccctgcgcgcccagggcatgcccatcgtcgccatcgactacctgccgccggagcggcgcgacgaggcgcgcgcgctcgctgcgcgcctgcgtagcgaaggctacgtgccgttcgtcagcaccccggcgctggactacctgggggtgagcgacgtcgaggtgcaaccg)。
55.2)pelan
‑
pa pcr扩增及pet28b表达载体构建:pcr扩增、引物对设计合成由苏州金唯智公司完成,选择ndei和hind iii酶切位点将pelan
‑
pa片段连接于pet28b载体上,制备pet28b
‑
pelan
‑
pa重组表达载体,并由苏州金唯智公司进行测序验证。
56.3)表达载体的转化:将步骤2)中的pet28b
‑
pelan
‑
pa重组表达载体导入到e.coli bl21(de3)感受态细胞中,并在含有终浓度为50μg/ml卡那霉素的lb固体培养基上进行阳性筛选。
57.4)工程菌株的扩大培养:挑取步骤3)中阳性单菌落于100ml含有终浓度50μg/ml卡那霉素的lb液体培养基中过夜培养,取5ml接种于500ml含有终浓度50μg/ml卡那霉素的lb液体培养基,于37℃,180r/min条件下进行扩增培养至od
600
为0.55
–
0.6。
58.5)pelan
‑
pa的诱导表达:向步骤4)扩增后的菌液中加入终浓度为1mmol/l的iptg,于20℃过夜诱导培养18h,在4℃、5000rpm条件下离心30min收集菌体。
59.6)细胞破碎:将步骤5)中收集到的菌体重悬于80ml细胞破碎液(20mm tris、300mm nacl、10mm imidazole、5%glycerol、30mg/ml的溶菌酶、罗氏蛋白酶抑制剂和dna酶)中,在冰浴条件下超声破碎,并于12000rmp离心收集上清液。
60.7)ni
‑
nta亲和层析柱分离:使用50ml的缓冲液1(20mm tris、300mm nacl、10mm imidazole、5%glycerol)平衡ni
‑
nta亲和层析柱;将步骤6)中收集的上清液通过ni
‑
nta亲和层析柱;用50ml的缓冲液2(20mm tris、500mm nacl、35mm imidazole、5%glycerol)充分洗去ni
‑
nta亲和层析柱上的杂蛋白;再用缓冲液3(20mm tris、500mm nacl、300mm imidazole、5%glycerol)将目标蛋白进行洗脱。
61.8)akta系统纯化:将120ml的凝胶层析柱superdex75 10/300gl利用缓冲液4(25mm tris、200mm nacl、5%glycerol)进行平衡,将步骤7)中ni
‑
nta柱纯化后的蛋白通过akta蛋白纯化系统进行凝胶层析实验(分子筛),收集分子筛峰图所对应的样品,并用sds
–
page验证纯化后的目标蛋白。
62.如图3所示,经过凝胶过滤层析(图3a)和sds
‑
page分析(图3b)可知,上述铜绿假单胞菌pa14生物被膜核心胞外多糖裂解酶pelan
‑
pa的制备方法成功获得重组pelan
‑
pa蛋白。
63.实施例2
64.一种荧光假单胞菌nbrc15842生物被膜核心胞外多糖裂解酶pelan
‑
pf的制备方法,包含以下步骤:
65.1)获取并设计细菌pelan
‑
pf基因组dna:从ncbi数据库下载获得荧光假单胞菌nbrc15842的全基因组序列(protein_id="ged73779.1"),并通过phyre2对其进行同源建模分析,发现其糖苷水解酶域位于第37
–
288位氨基酸(seq id no:3:pasvgfwyaeqpplqelaqfewavvepghmasadvatlrklgsqpfaylsvgefdgnraalakqalaqgaspvrnkawdsqvmdiatpawrehlfkrakalqdqgyaglfldtldsfqllpeadrepqrkalasflrelhsrlpnlklffnrgfevlgeldgvasavavesihagwdasakryrpvseadrtwlegelkplrarniplvaidylpanrreearklvrqlsqegfipvvttpdlnalsmstve),参照大肠杆菌遗传密码子频率表,通过在线密码子优化软件e.coli codon usage analyzer 2.1对序列进行稀有密码子偏好性优化,得到编码荧光假单胞菌nbrc15842生物被膜核心胞外多糖裂解酶pelan
‑
pf的核苷酸序列(seq id no:4:cccgccagcgtcgggttctggtacgccgagcagccacccttgcaggagctggcgcagttcgaatgggcagtggtcgagcccggccatatggccagcgccgatgtcgccactttgcgcaagctcggcagccagccgttcgcctacctgtcggtaggggagttcgacggcaaccgcgctgccctggccaagcaggccctggcccagggcgcgagccccgtgcgcaacaaggcctgggacagccaggtgatggacatcgccaccccggcctggcgcgaacacctgttcaagcgtgccaaggcgctgcaggaccagggctacgccggcctgttcctggacaccctggacagtttccagttgctccccgaagccgatcgcgaaccgcaacgcaaggccctggcctcgttcctgcgggaactgcacagccggctgcccaacctcaagctgttcttcaaccggggcttcgaggtactgggcgagctcgatggcgtcgcctcggccgtggcggtggaatccatccacgccggctgggatgcctcggccaagcgttaccgcccggtttccgaagccgaccgcacctggcttgaaggcgaactcaagccgctgcgcgcacgcaacatcccgctggtggccatcgattacctgccggccaaccgtcgggaagaggcacgcaagctggtccggcaattgagccaggaaggctttataccggtggtcaccaccccggatctgaacgccctgagcatgagcaccgtggaa)。
66.2)pelan
‑
pf pcr扩增及pet28b表达载体构建:pcr扩增、引物对设计合成由苏州金
唯智公司完成,选择ndei和hind iii酶切位点将pelan
‑
pf片段连接于pet28b载体上,制备重组表达载体,并由苏州金唯智公司进行测序验证。
67.3)表达载体的转化:将步骤2)中的表达载体导入到e.coli bl21(de3)感受态细胞中,并在含有终浓度为50μg/ml卡那霉素的lb固体培养基上进行阳性筛选。
68.4)工程菌株的扩大培养:挑取步骤3)中所述的阳性单菌落于100ml含有终浓度50μg/ml卡那霉素的lb液体培养基中过夜培养。取5ml接种于500ml含有终浓度50μg/ml卡那霉素的lb液体培养基,于37℃,180r/min条件下进行扩增培养至od
600
为0.55
–
0.6。
69.5)pelan
‑
pf的诱导表达:向步骤4)扩增后的菌液中加入终浓度为1mmol/l的iptg,于20℃过夜诱导培养18h,在4℃、5000rpm条件下离心30min收集菌体。
70.6)细胞破碎:将步骤5)中收集到的菌体重悬于80ml细胞破碎液(20mm tris、300mm nacl、10mm imidazole、5%glycerol,30mg/ml的溶菌酶,罗氏蛋白酶抑制剂和dna酶)中,在冰浴条件下超声破碎,并于12000rmp离心收集上清液。
71.7)ni
‑
nta亲和层析柱分离:使用50ml的缓冲液1(20mm tris、300mm nacl、10mm imidazole、5%glycerol)平衡ni
‑
nta亲和层析柱;将步骤6)中收集的上清液通过ni
‑
nta亲和层析柱;用50ml的缓冲液2(20mm tris、500mm nacl、35mm imidazole、5%glycerol)充分洗去ni
‑
nta亲和层析柱上的杂蛋白;再用缓冲液3(20mm tris、500mm nacl、300mm imidazole、5%glycerol)将目标蛋白进行洗脱。
72.8)akta系统纯化:将120ml的凝胶层析柱superdex75 10/300gl利用缓冲液4(25mm tris、200mm nacl、5%glycerol)进行平衡,将步骤7)中ni
‑
nta柱纯化后的蛋白通过akta蛋白纯化系统进行凝胶层析实验(分子筛),收集分子筛峰图所对应的样品,并用sds
–
page验证纯化后的目标蛋白。
73.如图4所示,经过凝胶过滤层析(图4a)和sds
‑
page分析(图4b)可知,上述荧光假单胞菌nbrc15842生物被膜核心胞外多糖裂解酶pelan
‑
pf的制备方法成功获得重组pelan
‑
pf蛋白。
74.实施例3
75.一种恶臭假单胞菌jbc17生物被膜核心胞外多糖裂解酶pelan
‑
pp的制备方法,包含以下步骤:
76.1)获取并设计细菌pelan
‑
pp基因组dna:从ncbi数据库下载获得恶臭假单胞菌jbc17的全基因组序列(id:awy42675.1),并通过phyre2对其进行同源建模分析,发现其糖苷水解酶域位于第38
–
289位氨基酸(seq id no:5:pssvsfwyadepplaelaqfawtvvepghmtaadvktlrklgsepfaylsvgefdgskaditkagltkavspvrndswnsqvmdltspawrdhllgrakqlqaqgygglfldtldsftllpqaaqeaqraglasflrelhkqqpqlklffnrgfevlpeldgvaaavafeslyagwdaaakryrpvpeadrqwllgqlqplrakgiplvaidylpperrdearklakrlrdegfipfistpdlnsmgistve),参照大肠杆菌遗传密码子频率表,通过在线密码子优化软件e.coli codon usage analyzer 2.1对序列进行稀有密码子偏好性优化,得到编码恶臭假单胞菌jbc17生物被膜核心胞外多糖裂解酶pelan
‑
pp的核苷酸序列(seq id no:6:ccttccagtgtgtccttctggtatgccgacgagccgccgctggccgagctggcgcagttcgcctggacggtggtcgagccggggcatatgacggcggcggatgtcaaaaccctgcgcaagctgggcagcgagccgttcgcctatctgtcggtcggcgagttcgacggatccaaggctgacatcaccaaagcgggcctcaccaaggccgtttccccggtgcgcaacgactcgtggaacagtcaggtcatggacctcacttctccagcc
tggcgcgaccacttgctgggtcgggccaagcaattgcaggcccagggctatggcggcctgttcctcgataccctcgacagtttcacgctgttgccgcaagccgcgcaggaagcgcagcgcgcggggctggccagtttcctgcgcgaactgcacaagcagcagcctcagctgaagctgttctttaaccgtggctttgaagtgttgcccgagctcgacggcgtggcggcagcggtggcattcgagtcgctgtatgccggctgggatgcggcggccaagcgttatcgcccggtgccggaagccgatcgccagtggctgctgggccagttgcaaccgttgcgcgccaagggcattccactggtggccatcgattatttgccgccggagcgtcgcgacgaggcccgcaagctggccaagcgtctgcgtgacgaaggcttcattcctttcatcagcacccctgacctcaactcgatgggcatcagcaccgtcgaa)。
77.2)pelan
‑
pp pcr扩增及pet28b表达载体构建:pcr扩增、引物对设计合成由苏州金唯智公司完成,选择ndei和hind iii酶切位点将pelan
‑
pp片段连接于pet28b载体上,制备重组表达载体,并由苏州金唯智公司进行测序验证。
78.3)表达载体的转化:将步骤2)中的表达载体导入到e.coli bl21(de3)感受态细胞中,并在含有终浓度为50μg/ml卡那霉素的lb固体培养基上进行阳性筛选。
79.4)工程菌株的扩大培养:挑取步骤3)中所述的阳性单菌落于100ml含有终浓度50μg/ml卡那霉素的lb液体培养基中过夜培养。取5ml接种于500ml含有终浓度50μg/ml卡那霉素的lb液体培养基,于37℃,180r/min条件下进行扩增培养至od
600
为0.55
–
0.6。
80.5)pelan
‑
pp的诱导表达:向步骤4)扩增后的菌液中加入终浓度为1mmol/l的iptg,于20℃过夜诱导培养18h,在4℃、5000rpm条件下离心30min收集菌体。
81.6)细胞破碎:将步骤5)中收集到的菌体重悬于80ml细胞破碎液(20mm tris、300mm nacl、10mm imidazole、5%glycerol,30mg/ml的溶菌酶,罗氏蛋白酶抑制剂和dna酶)中,在冰浴条件下超声破碎,并于12000rmp离心收集上清液。
82.7)ni
‑
nta亲和层析柱分离:使用50ml的缓冲液1(20mm tris、300mm nacl、10mm imidazole、5%glycerol)平衡ni
‑
nta亲和层析柱;将步骤6)中收集的上清液通过ni
‑
nta亲和层析柱;用50ml的缓冲液2(20mm tris、500mm nacl、35mm imidazole、5%glycerol)充分洗去ni
‑
nta亲和层析柱上的杂蛋白;再用缓冲液3(20mm tris、500mm nacl、300mm imidazole、5%glycerol)将目标蛋白进行洗脱。
83.8)akta系统纯化:将120ml的凝胶层析柱superdex75 10/300gl利用缓冲液4(25mm tris、200mm nacl、5%glycerol)进行平衡,将步骤7)中ni
‑
nta柱纯化后的蛋白通过akta蛋白纯化系统进行凝胶层析实验(分子筛),收集分子筛峰图所对应的样品,并用sds
–
page验证纯化后的目标蛋白。
84.如图5所示,经过凝胶过滤层析(图5a)和sds
‑
page分析(图5b)可知,上述恶臭假单胞菌jbc17生物被膜核心胞外多糖裂解酶pelan
‑
pp的制备方法成功获得重组pelan
‑
pp蛋白。
85.实施例4
86.将实施例1至3制备的细菌生物被膜核心胞外多糖裂解酶pelan
‑
pa、pelan
‑
pf和pelan
‑
pp用于抑制铜绿假单胞菌pa14生物被膜,步骤如下:
87.1)铜绿假单胞菌pa14的培养:铜绿假单胞菌pa14在37℃,200r/min下过夜培养。
88.2)生物被膜的形成:将步骤1)中培养的菌液标准化至od
600
=0.5,然后用lb液体培养基1:100稀释;取95μl稀释液及5μl浓度为5μm的pelan
‑
pa或pelan
‑
pf或pelan
‑
pp,并取100μl加至无菌96孔板中作为阳性对照,在25℃下静置培养24h形成生物被膜。
89.3)生物被膜量测定:选用结晶紫法测定生物被膜的形成,即用无菌水彻底清洗孔板,洗去浮游细胞,然后用0.1%(w/v)150μl结晶紫染色10min,用水冲洗掉结晶紫,剩余的部分用95%(v/v)150μl乙醇溶解10min,最后在od595下测定其吸光度。
90.如图6所示,经细菌生物被膜核心胞外多糖裂解酶pelan
‑
pa、pelan
‑
pf和pelan
‑
pp预处理后的铜绿假单胞菌pa14的生物被膜形成量显著低于对照组,证明细菌生物被膜核心胞外多糖裂解酶pelan可作为一种绿色、高效的生物被膜清除剂。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1