一种3-酰基吡咯类化合物的合成方法与流程
一种3
‑
酰基吡咯类化合物的合成方法
技术领域
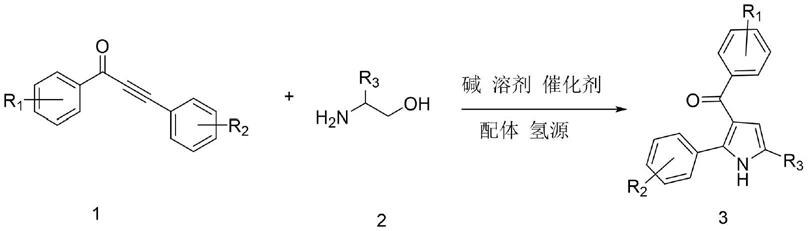
1.本发明涉及医药化工合成技术领域,具体为一种3
‑
酰基吡咯类化合物的合成方法。
背景技术:
2.吡咯结构是众多杂环化合物中最常见的骨架之一,作为一类重要的五元氮杂环化合物,其具有多种生物活性和药理活性,常出现在天然产物和一些上市药物结构中。对于多取代吡咯化合物,3
‑
酰基吡咯结构在医药和功能性材料方面有着广泛应用,是吡咯家族中的明星分子。例如,有效的组蛋白去乙酰化酶抑制剂,hiv
‑
1转录酶和cox
‑
1/cox
‑
2环氧合酶。此外,一些3
‑
酰基吡咯衍生物是有效的降胆固醇药物,抑制hmg
‑
coa还原酶(jannordmann and thomas j.j.m
ü
ller,org biomol chem,2013,11(38),6556
‑
6561)。此外,酰基的存在,有利于各种衍生化合成,可作为重要的合成子参与制备复杂的功能性分子。
3.由于3
‑
酰基吡咯类化合物具有独特的生物活性和广泛的用途,3
‑
酰基吡咯化合物的高效合成一直是有机合成化学中的研究热点。近年来,针对3
‑
酰基吡咯化合物的合成已经建立了一些有效的合成方法。例如:ibx介导的n
‑
羟基烷基烯胺的分子内氧化环化反应,生成相应的2,3
‑
二取代吡咯和吡啶产物(peng gao,huai
‑
juan chen,zi
‑
jing bai,mi
‑
na zhao,desuo yang,juanwang,ning wang,lele du,and zheng
‑
hui guan,j.org.chem,2020,85(12),7939
‑
7951)。二氯化钯和二(1
‑
金刚烷基)苄基溴化膦催化的一锅法三组分酸性氯化物、末端炔烃和氨基乙醛二乙缩醛合成2取代酰基吡咯(jannordmann and thomas j.j.m
ü
ller,org biomol chem,2013,11(38),6556
‑
6561)。β
‑
氨基烯酮与α
‑
氨基酮通过两步反应得到2
‑
或3
‑
酰基吡咯(angelalberola*,jose ei.andres,alfonso gonadlez,rafael pedrosa,and martina yicente,heterocycles,1989,29(10),1983
‑
91)。上述反应能够有效得到3
‑
酰基吡咯衍生物,但是大多存在一些不足。例如:原料不易得,反应过程繁琐,使用毒性试剂和贵重金属,反应条件较为苛刻等,从而难以应用于工业生产。
技术实现要素:
4.本发明的目的在于提供一种3
‑
酰基吡咯类化合物的合成方法,以解决上述背景技术中提出的问题。
5.为实现上述目的,本发明提供如下技术方案:一种3
‑
酰基吡咯类化合物的合成方法,包括以下步骤:
6.在反应器中,加入化合物1和化合物2,催化剂,配体,氢源,碱和适量溶剂,在80℃氮气保护下搅拌1.5小时;然后升温到150℃反应18小时,反应结束后冷却至室温,稀释反应液,过滤,减压蒸馏得粗产物,经柱层析提纯得到3
‑
酰基吡咯类化合物;
7.上述合成方法所涉及的反应方程式如下:
[0008][0009]
所述的化合物1是指具有式(1)结构的化合物:α,β
‑
不饱和炔酮;所述的化合物2是指具有式(2)结构的化合物:α
‑
氨基醇;
[0010][0011]
其中,r1为甲基、甲氧基、三氟甲基、氰基、卤素取代基或氢;r2为相同或者不相同的多取代苯基、苯基;r3为甲基、乙基、异丙基、苯基或氢。
[0012]
优选的,所述化合物1与化合物2的摩尔比为1(mmol):1(mmol)。
[0013]
优选的,所述配体是指4
‑
甲基
‑
1,10菲罗啉;4
‑
甲基
‑
1,10菲罗啉的加入量与化合物1的摩尔比为1:10。
[0014]
优选的,所述氢源是指乙醇;乙醇的加入量与化合物1的摩尔比为2:1。
[0015]
优选的,所述催化剂是指十二羰基三钌;十二羰基三钌的加入量与化合物1的摩尔比为0.02:1。
[0016]
优选的,所述溶剂为叔戊醇,叔戊醇的加入量与化合物1的配比为1(ml):0.1(mmol)。
[0017]
优选的,所述碱为碳酸钾;碳酸钾的加入量与化合物1的摩尔比为1:1。
[0018]
优选的,所述的柱层析提纯所用的洗脱液为石油醚:乙酸乙酯的体积比为(5~20):1的混合溶剂。
[0019]
优选的,所述反应器为史兰克管(schlenk管)。
[0020]
与现有技术相比,本发明的有益效果是:本发明提出的一种3
‑
酰基吡咯类化合物的合成方法,以α
‑
氨基醇类化合物和α,β
‑
不饱和炔酮为原料,通过串联式一锅法合成3
‑
酰基吡咯类化合物,具有合成步骤简单、原料易得、合成操作安全、官能团兼容性好等优点。
附图说明
[0021]
图1为本发明实施例1所得产物3a的氢谱图;
[0022]
图2为本发明实施例1所得产物3a的碳谱图;
[0023]
图3为本发明实施例2所得产物3b的氢谱图;
[0024]
图4为本发明实施例2所得产物3b的碳谱图;
[0025]
图5为本发明实施例3所得产物3c的氢谱图;
[0026]
图6为本发明实施例3所得产物3c的碳谱图;
[0027]
图7为本发明实施例4所得产物3d的氢谱图;
[0028]
图8为本发明实施例4所得产物3d的碳谱图;
[0029]
图9为本发明实施例5所得产物3e的氢谱图;
[0030]
图10为本发明实施例5所得产物3e的碳谱图。
具体实施方式
[0031]
下面将结合本发明实施例中的附图,对本发明实施例中的技术方案进行清楚、完整地描述,显然,所描述的实施例仅仅是本发明一部分实施例,而不是全部的实施例。基于本发明中的实施例,本领域普通技术人员在没有做出创造性劳动前提下所获得的所有其他实施例,都属于本发明保护的范围。
[0032]
实施例1
[0033]
在schlenk管中加入0.3毫摩尔间氟苯酮炔基苯,0.3毫摩尔2
‑
氨基
‑1‑
丁醇,0.006毫摩尔十二羰基三钌,0.03毫摩尔4
‑
甲基
‑
1,10菲罗琳,0.6毫摩尔乙醇,0.3毫摩尔碳酸钾和3毫升叔戊醇,氮气保护条件下80℃反应1.5小时,随后,升温到150℃反应18小时,冷却至室温,用乙酸乙酯稀释反应液,过滤,减压蒸馏除去溶剂。柱层析分离纯化,得到目标产物3a,所用柱层析洗脱液为体积比为15:1的石油醚:乙酸乙酯混合溶剂,产率77%。
[0034]
所得产物3a的氢谱和碳谱图分别如图1和图2所示,结构表征数据如下:
[0035]1h nmr(400mhz,cdcl3)δ8.51(s,1h),7.41(dd,j=7.6,1.6hz,1h),7.33(dt,j=9.4,2.0hz,1h),7.29
–
7.24(m,2h),7.19
–
7.09(m,4h),6.99(td,j=8.3,2.6hz,1h),6.20(d,j=2.8hz,1h),2.56(q,j=7.6hz,2h),1.20(t,j=7.5hz,3h).
[0036]
13
c nmr(101mhz,cdcl3):δ190.08,162.42,159.97,140.84,135.55,133.55,131.02,128.33,127.36,126.80,124.24,118.98,117.27,117.06,115.36,115.14,108.21,19.45,12.21.
[0037]
ms(ei,m/z):293[m]
+
.
[0038]
hrms(esi):calcd.for c19h16fno[m+h]
+
:294.1289;found:294.1284.
[0039]
根据以上数据推断所得产物的结构如下式所示:
[0040][0041]
实施例2
[0042]
在schlenk管中加入0.3毫摩尔对甲基苯酮炔基苯,0.3毫摩尔2
‑
氨基
‑1‑
丁醇,0.006毫摩尔十二羰基三钌,0.03毫摩尔4
‑
甲基
‑
1,10菲罗琳,0.6毫摩尔乙醇,0.3毫摩尔碳酸钾和3毫升叔戊醇,氮气保护条件下80℃反应1.5小时,随后,升温到150℃反应18小时,冷却至室温,用乙酸乙酯稀释反应液,过滤,减压蒸馏除去溶剂。柱层析分离纯化,得到目标产物3b,所用柱层析洗脱液为体积比为15:1的石油醚:乙酸乙酯混合溶剂,产率70%。
[0043]
所得产物3b的氢谱和碳谱图分别如图3和图4所示,结构表征数据如下:
[0044]1h nmr(400mhz,cdcl3)δ8.27(s,1h),7.31(d,j=4.8hz,2h),7.24
–
7.07(m,5h),
7.03(d,j=7.4hz,1h),6.94(t,j=7.4hz,1h),6.09(d,j=2.9hz,1h),2.53(q,j=7.6hz,2h),2.28(s,3h),1.17(t,j=7.5hz,3h).
[0045]
13
c nmr(101mhz,cdcl3):δ193.17,139.76,135.60,135.08,133.20,131.06,129.41,128.28,127.48,127.40,127.04,126.79,123.72,120.59,108.29,19.44,18.82,12.14.
[0046]
ms(ei,m/z):289[m]
+
.
[0047]
hrms(esi):calcd.for c20h19no[m+h]
+
:290.1539;found:290.1538.
[0048]
根据以上数据推断所得产物的结构如下式所示:
[0049][0050]
实施例3
[0051]
在schlenk管中加入0.3毫摩尔苯酮炔基苯,0.3毫摩尔2
‑
氨基
‑1‑
丁醇,0.006毫摩尔十二羰基三钌,0.03毫摩尔4
‑
甲基
‑
1,10菲罗琳,0.6毫摩尔乙醇,0.3毫摩尔碳酸钾和3毫升叔戊醇,氮气保护条件下80℃反应1.5小时,随后,升温到150℃反应18小时,冷却至室温,用乙酸乙酯稀释反应液,过滤,减压蒸馏除去溶剂。柱层析分离纯化,得到目标产物3c,所用柱层析洗脱液为体积比为15:1的石油醚:乙酸乙酯混合溶剂,产率72%。
[0052]
所得产物3c的氢谱和碳谱图分别如图5和图6所示,结构表征数据如下:
[0053]1h nmr(400mhz,cdcl3)δ8.30(s,1h),7.68(d,j=7.3hz,2h),7.31(d,j=7.1hz,3h),7.25
–
7.12(m,5h),6.22(d,j=2.4hz,1h),2.59(q,j=7.6hz,2h),1.21(t,j=7.5hz,3h).
[0054]
13
c nmr(101mhz,cdcl3):δ192.58,139.74,136.07,134.23,132.28,131.41,129.63,128.36,128.29,127.77,127.70,120.49,109.55,20.57,13.30.
[0055]
ms(ei,m/z):275[m]
+
.
[0056]
hrms(esi):calcd.for c19h17no[m+h]
+
:276.1383;found:276.1380.
[0057]
根据以上数据推断所得产物的结构如下式所示:
[0058][0059]
实施例4
[0060]
在schlenk管中加入0.3毫摩尔对三氟甲基苯酮炔基苯,0.3毫摩尔2
‑
氨基
‑1‑
丁醇,0.006毫摩尔十二羰基三钌,0.03毫摩尔4
‑
甲基
‑
1,10菲罗琳,0.6毫摩尔乙醇,0.3毫摩
尔碳酸钾和3毫升叔戊醇,氮气保护条件下80℃反应1.5小时,随后,升温到150℃反应18小时,冷却至室温,用乙酸乙酯稀释反应液,过滤,减压蒸馏除去溶剂。柱层析分离纯化,得到目标产物3d,所用柱层析洗脱液为体积比为15:1的石油醚:乙酸乙酯混合溶剂,产率80%。
[0061]
所得产物3d的氢谱和碳谱图分别如图7和图8所示,结构表征数据如下:
[0062]1h nmr(400mhz,cdcl3)δ8.54(s,1h),7.68(d,j=8.1hz,2h),7.43(d,j=8.2hz,2h),7.28
–
7.21(m,2h),7.15
–
7.09(m,3h),6.20(d,j=2.8hz,1h),2.56(q,j=7.6hz,2h),1.20(t,j=7.5hz,3h).
[0063]
13
c nmr(101mhz,cdcl3):δ191.29,142.96,137.05,134.78,132.74,132.41,131.93,129.62,128.56,128.29,128.03,124.76,120.04,109.21,20.51,13.24.
[0064]
ms(ei,m/z):343[m]
+
.
[0065]
hrms(esi):calcd.for c20h16f3no[m+h]
+
:344.1257;found:344.1255.
[0066]
根据以上数据推断所得产物的结构如下式所示:
[0067][0068]
实施例5
[0069]
在schlenk管中加入0.3毫摩尔对甲基苯酮炔基苯,0.3毫摩尔亮氨醇,0.006毫摩尔十二羰基三钌,0.03毫摩尔4
‑
甲基
‑
1,10菲罗琳,0.6毫摩尔乙醇,0.3毫摩尔碳酸钾和3毫升叔戊醇,氮气保护条件下80℃反应1.5小时,随后,升温到150℃反应18小时,冷却至室温,用乙酸乙酯稀释反应液,过滤,减压蒸馏除去溶剂。柱层析分离纯化,得到目标产物3e,所用柱层析洗脱液为体积比为15:1的石油醚:乙酸乙酯混合溶剂,产率65%。
[0070]
所得产物3e的氢谱和碳谱图分别如图1和图2所示,结构表征数据如下:
[0071]1h nmr(400mhz,cdcl3)δ8.27(s,1h),7.33(d,j=7.3hz,2h),7.22(d,j=7.5hz,1h),7.17
–
7.08(m,4h),7.03(d,j=7.5hz,1h),6.95(t,j=7.4hz,1h),6.07(d,j=2.8hz,1h),2.35(d,j=7.1hz,2h),2.28(s,3h),1.79(dt,j=13.5,6.7hz,1h),0.87(d,j=6.6hz,6h).
[0072]
13
c nmr(101mhz,cdcl3):δ193.25,135.42,135.43,135.13,131.09,130.81,129.43,128.32,127.57,127.40,127.06,126.77,123.75,120.67,110.09,76.32,76.00,75.68,35.67,27.87,21.36,18.87.
[0073]
ms(ei,m/z):317[m]
+
.
[0074]
hrms(esi):calcd.for c22h23no[m+h]
+
:318.1852;found:318.1848.
[0075]
根据以上数据推断所得产物的结构如下式所示:
[0076][0077]
本发明未详述之处,均为本领域技术人员的公知技术。
[0078]
对于本领域技术人员而言,显然本发明不限于上述示范性实施例的细节,而且在不背离本发明的精神或基本特征的情况下,能够以其他的具体形式实现本发明。因此,无论从哪一点来看,均应将实施例看作是示范性的,而且是非限制性的,本发明的范围由所附权利要求而不是上述说明限定,因此旨在将落在权利要求的等同要件的含义和范围内的所有变化囊括在本发明内。不应将权利要求中的任何附图标记视为限制所涉及的权利要求。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1