一种海洋真菌杂色曲霉M-7-SW9、混源萜类化合物及其提取方法和应用
一种海洋真菌杂色曲霉m
‑7‑
sw9、混源萜类化合物及其提取方法和应用
技术领域
1.本发明属于微生物医药技术领域,具体涉及一种海洋真菌杂色曲霉m
‑7‑
sw9、混源萜类化合物及其提取方法和应用。
背景技术:
2.水产养殖业细菌性病害的威胁是世界性的,几乎侵害所有的水产养殖动物,不仅造成重大的经济损失,也为水产品质量安全埋下了隐患。据水产养殖动物病害监测结果显示,近年来水产养殖病害种类高达200余种,其中细菌性疾病占48%以上。在细菌性水产动物疾病中,由弧菌属细菌引起的弧菌病是最主要的细菌性疾病,该病发生范围广、致死率高,且能够引起人畜共患,是水产养殖病害防治领域的主要难题之一。
3.近年来,随着水产养殖规模的发展和水产养殖过程中传统型抗生素的大量使用,水产养殖动物病原菌和养殖环境中细菌的多重耐药现象日趋普遍,并且化学合成药物的大量使用对水体和土壤带来了严重的污染,对生物和人类健康都造成了直接和潜在的危害。
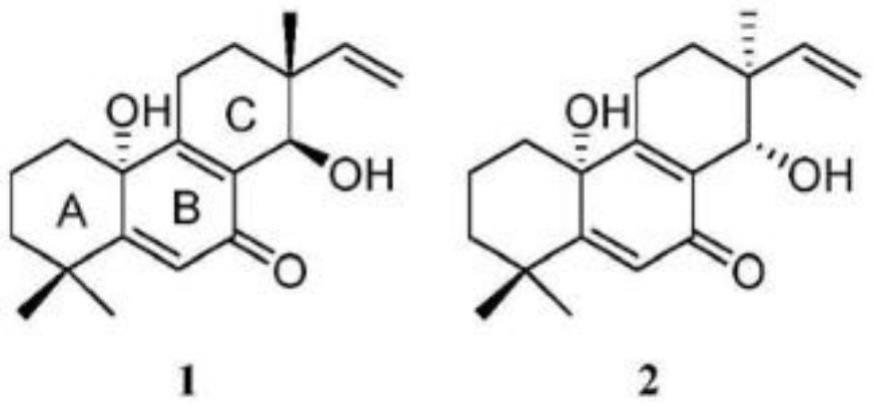
4.与水产养殖业中使用的传统抗生素和化学合成药物相比,海洋生物源天然药物具有安全性高、针对性强、活性显著、对环境友好且能够解决目前大部分水产病害细菌对传统抗生素的耐药性问题,为解决目前水产养殖过程中的细菌性病害防治问题提供了新的思路。例如,中国专利cn201810906911.9公开了一种海洋真菌温特曲霉来源的降二萜类化合物,化合物1和化合物2,其结构式如下:
[0005][0006]
然而,上述化合物对于迟缓爱德华氏菌和哈氏弧菌的最低抑制浓度(mic值)均为8.0μg/ml,对水产病害细菌的抑制活性不够高。
技术实现要素:
[0007]
有鉴于此,本发明的目的在于提供一种海洋真菌杂色曲霉m
‑7‑
sw9、混源萜类化合物及其提取方法和应用,从所述海洋真菌杂色曲霉m
‑7‑
sw9中提取出来的混源萜类化合物对水产病害细菌,尤其是对迟缓爱德华氏菌和哈氏弧菌的抑制活性高。
[0008]
为了实现上述发明目的,本发明提供以下技术方案:
[0009]
本发明提供了一种海洋真菌杂色曲霉(aspergillus versicolor)m
‑7‑
sw9,所述海洋真菌杂色曲霉m
‑7‑
sw9的保藏编号为cctcc no:m 2021454。
[0010]
本发明提供了一种混源萜类化合物,具有式i所示的结构:
[0011][0012]
本发明提供了上述技术方案所述混源萜类化合物的提取方法,包括以下步骤:
[0013]
(1)将权利要求1所述的海洋真菌杂色曲霉m
‑7‑
sw9在pdb液体培养基上进行发酵培养,采用极性有机溶剂对得到的发酵产物进行提取,得到发酵粗提物;
[0014]
(2)将所述发酵粗提物进行正相硅胶柱层析分离,得到洗脱组分;所述正相硅胶柱层析分离的洗脱方式为梯度洗脱,所述梯度洗脱采用的洗脱剂为低极性溶剂
‑
高极性溶剂;所述低极性溶剂
‑
高极性溶剂中低极性溶剂和高极性溶剂的体积比为50~0:1;
[0015]
(3)将所述洗脱组分进行反相硅胶柱层析分离,得到粗产物;所述反相硅胶柱层析分离的洗脱方式为梯度洗脱,所述梯度洗脱采用的洗脱剂为水
‑
醇溶剂,所述水
‑
醇溶剂中水和醇的体积比为4~0:1;
[0016]
(4)将所述粗产物进行纯化,得到具有式i所示结构的混源萜类化合物;所述纯化包括依次进行的凝胶柱层析和薄层层析,或,高效液相色谱纯化;
[0017]
所述凝胶柱层析采用的洗脱剂为二氯甲烷
‑
醇溶剂;所述二氯甲烷
‑
醇溶剂中二氯甲烷和醇的体积比为2~0:1;
[0018]
所述薄层层析采用的展开剂为石油醚
‑
乙酸乙酯混合溶剂,所述石油醚
‑
乙酸乙酯混合溶剂中石油醚和乙酸乙酯的体积比为2:1;
[0019]
所述高效液相色谱纯化的流动相为水
‑
甲醇混合溶剂,所述水
‑
甲醇混合溶剂中甲醇和水的体积比为1:1。
[0020]
优选的,所述pdb液体培养基以水溶剂,包括土豆粉和/或小麦粉200~300g/l,葡萄糖20~30g/l,蛋白胨5~8g/l,酵母浸粉3~5g/l和氯化钠35~40g/l。
[0021]
优选的,所述发酵培养的温度为25~30℃,时间为7~14天。
[0022]
优选的,所述极性有机溶剂包括二氯甲烷、乙酸乙酯和低级醇中的一种或几种。
[0023]
优选的,所述提取的温度为20~40℃,时间为3~7天。
[0024]
本发明提供了上述技术方案所述的混源萜类化合物或上述技术方案所述提取方法得到的混源萜类化合物在制备抗水产病害细菌药物中的应用。
[0025]
本发明提供了一种海洋真菌杂色曲霉(aspergillus versicolor)m
‑7‑
sw9,其特征在于,所述海洋真菌杂色曲霉m
‑7‑
sw9的保藏编号为cctcc no:m 2021454。本发明提供的海洋真菌杂色曲霉m
‑7‑
sw9中能够提取出具有式i所示结构的混源萜类化合物,所述混源萜类化合物对水产病害细菌,尤其是对迟缓爱德华氏菌和哈氏弧菌的抑制活性高。
[0026]
本发明提供了一种混源萜类化合物,具有式i所示的结构。本发明提供的混源萜类化合物对水产病害细菌的抑制活性高,尤其是对迟缓爱德华氏菌和哈氏弧菌的抑制活性高;而且,与水产养殖业中使用的传统抗生素和化学合成药物相比,本发明提供的混源萜类化合物为海洋生物源天然药物,为开发海洋生物源天然药物以解决水产养殖过程中的细菌性病害防治问题提供了新的思路。如实施例测试结果所示,本发明提供的混源萜类化合物对于迟缓爱德华氏菌和哈氏弧菌的最低抑菌浓度(mic值)均为1.0μg/ml;氯霉素对迟缓爱
德华氏菌的mic值分别为2.3μg/ml,对哈氏弧菌的mic值为2.2μg/ml;说明,本发明提供的混源萜类化合物对迟缓爱德华氏菌和哈氏弧菌的抑制活性高于氯霉素、化合物1和化合物2。
[0027]
本发明提供了上述技术方案所述混源萜类化合物的提取方法。本发明提供的提取方法操作简单,成本低,适宜规模化生产。
[0028]
生物保藏信息
[0029]
海洋真菌杂色曲霉(aspergillus versicolor)m
‑7‑
sw9,保藏日期为2021年04月25日,保藏地点为中国典型培养物保藏中心,具体地址为中国.武汉.武汉大学,保藏编号为cctccno:m 2021454。
具体实施方式
[0030]
本发明提供了一种海洋真菌杂色曲霉(aspergillus versicolor)m
‑7‑
sw9,所述海洋真菌杂色曲霉m
‑7‑
sw9的保藏编号为cctcc no:m 2021454。在本发明中,所述海洋真菌杂色曲霉m
‑7‑
sw9分离采集自海水样品中;所述海水样品优选为山东省烟台市养马岛附近海域水深0.5~3m处采集的海水样品。在本发明中,所述海洋真菌杂色曲霉m
‑7‑
sw9的分离方法优选包括以下步骤:将海水样品用无菌蒸馏水稀释后涂抹到pda培养基上,然后置于真菌培养箱中静置培养,待所述pda培养基表面长出真菌菌落后,将各真菌菌落分别转移至pda培养基上继续培养,重复转移直至所述pda培养基上的菌落为单一菌株的菌落,然后进行菌株鉴定。在本发明中,所述pda培养基在使用前优选进行消毒处理,本发明对于所述消毒的方式没有特殊限定,采用本领域技术人员熟知的培养基的消毒方法即可。在本发明中,所述稀释的倍数优选为5~20倍,更优选为10倍。在本发明中,所述静置培养的温度优选为25~30℃,更优选为28℃。在本发明中,所述海洋真菌杂色曲霉m
‑7‑
sw9在pda培养基上形成的菌落为白色气生菌丝,生长的孢子为灰绿色孢子。在本发明中,所述菌种鉴定优选采用dp305试剂盒(tiangen生物技术公司)进行,通过提取并克隆菌种核糖体rdna基因转录间隔序列(its1
‑
5.8s
‑
its2全长序列),然后进行基因测序,得到菌种序列后在genbank中与已知菌株的基因序列进行比较,所述菌株的基因序列如seq id no.1所示,所述菌株的dna序列与真菌杂色曲霉aspergillus versicolor(accessionno.mh911415.1)具有99%的同源性,因此,将所述的菌株命名为海洋真菌杂色曲霉(aspergillus versicolor)m
‑7‑
sw9。
[0031]
本发明提供了一种混源萜类化合物,具有式i所示的结构:
[0032][0033]
本发明提供了上述技术方案所述混源萜类化合物的提取方法,包括以下步骤:
[0034]
将上述技术方案所述的海洋真菌杂色曲霉m
‑7‑
sw9在pdb液体培养基上进行发酵培养,然后采用极性有机溶剂对得到的发酵产物进行提取,得到发酵粗提物;
[0035]
将所述发酵粗提物进行正相硅胶柱层析分离,得到洗脱组分;所述正相硅胶柱层析分离的洗脱方式为梯度洗脱,所述梯度洗脱采用的洗脱剂为低极性溶剂
‑
高极性溶剂;所
述低极性溶剂和高极性溶剂的体积比为50~0:1;
[0036]
将所述洗脱组分进行反相硅胶柱层析分离,得到粗产物;所述反相硅胶柱层析分离的洗脱方式为梯度洗脱,所述梯度洗脱采用的洗脱剂为水
‑
醇溶剂;所述水
‑
醇溶剂中水和醇的体积比为4~0:1;
[0037]
将所述粗产物进行纯化,得到具有式i所示结构的混源萜类化合物;所述纯化包括依次进行的凝胶柱层析和薄层层析,或,高效液相色谱纯化;
[0038]
所述凝胶柱层析采用的洗脱剂为二氯甲烷
‑
醇溶剂;所述二氯甲烷
‑
醇溶剂中二氯甲烷和醇的体积比为2~0:1;
[0039]
所述薄层层析采用的展开剂为石油醚
‑
乙酸乙酯混合溶剂,所述石油醚
‑
乙酸乙酯混合溶剂中石油醚和乙酸乙酯的体积比为2:1;
[0040]
所述高效液相色谱纯化的流动相为水
‑
甲醇混合溶剂,所述水
‑
甲醇混合溶剂中甲醇和水的体积比为1:1。
[0041]
本发明将上述技术方案所述的海洋真菌杂色曲霉m
‑7‑
sw9在pdb液体培养基上进行发酵培养,然后采用极性有机溶剂对得到的发酵产物进行提取,得到发酵粗提物。
[0042]
在本发明中,所述pdb液体培养基优选以水为溶剂,优选包括:土豆粉和/或小麦粉200~300g/l,优选为250g/l;葡萄糖20~30g/l,优选为25g/l;蛋白胨5~8g/l,优选为6~7g/l;酵母浸粉3~5g/l,优选为4~4.5g/l;氯化钠35~40g/l,优选为36~38g/l。在本发明中,所述发酵培养的温度优选为25~30℃,更优选为28℃;所述发酵培养的时间优选为7~14天,更优选为14天。
[0043]
在本发明中,所述发酵培养的步骤优选包括:取生长于平板培养基中的海洋真菌杂色曲霉m
‑7‑
sw9接种于第一pdb液体培养基中第一发酵培养,得到菌液;将所述菌液接种到第二pdb液体培养基中进行第二发酵培养。在本发明中,所述第一pdb液体培养基和第二pdb液体培养基在使用前优选进行行消毒处理,本发明对于所述消毒的方式没有特殊限定,采用本领域技术人员熟知的平板培养基的消毒方法即可。在本发明中,所述平板培养基的尺寸优选为1.5cm
×
1.5cm;所述第一pdb液体培养基的体积优选为200~300ml,更优选为250ml;所述第一发酵培养的温度优选为25~30℃,更优选为28℃;所述第一发酵培养时间优选为7~14天,更优选为7天;所述第一发酵培养优选在摇床上进行,所述摇床的转速优选为150~250r/min,更优选为200r/min。在本发明中,所述第二pdb液体培养基的体积优选为250~350l,更优选为300l;所述第二发酵培养的温度优选为25~30℃,更优选为28℃;所述第二发酵培养时间优选为7~14天,更优选为7天;所述第二发酵培养优选在发酵罐中进行;所述发酵罐中第二pdb液体培养基的填充体积分数优选为50~70%,更优选为60%。
[0044]
在本发明中,所述极性有机溶剂优选包括二氯甲烷、乙酸乙酯和低级醇中的一种或几种,所述低级醇优选包括甲醇、乙醇、丙醇和异丙醇中一种或几种。在本发明中,所述提取的温度优选为20~40℃,更优选为25~30℃,所述提取的时间优选为3~7天,更优选为4~5天。
[0045]
所述提取后,本发明优选还包括将所述提取的体系进行萃取,将所得有机相进行浓缩,得到发酵粗提物。在本发明中,所述萃取采用的萃取剂优选包括乙酸乙酯、石油醚或正丁醇;所述萃取的次数优选为3~5次,更优选为4次。本发明对于所述浓缩没有特殊限定,采本领域技术人员熟知的浓缩方式即可,具体如利用旋转蒸发仪进行减压加热浓缩,浓缩
至无萃取剂残留即可。
[0046]
得到发酵粗提物后,本发明将所述发酵粗提物进行正相硅胶柱层析分离,得到洗脱组分。在本发明中,所述正相硅胶柱层析分离的洗脱方式为梯度洗脱,所述梯度洗脱采用的洗脱剂为低极性溶剂
‑
高极性溶剂;所述低极性溶剂优选包括石油醚和/或二氯甲烷;所述高极性溶剂优选包括乙酸乙酯、乙醇、丙醇和异丙醇中的一种或几种;所述低极性溶剂
‑
高极性溶剂中低极性溶剂和高极性溶剂的体积比为50~0:1;所述梯度洗脱过程中低极性溶剂和高极性溶剂的体积比依次分别为50:1、40:1、30:1、20:1、10:1和0:1,所述低极性溶剂和高极性溶剂的体积比为10:1的组分即为所述洗脱组分。
[0047]
得到所述洗脱组分后,本发明将所述洗脱组分进行反相硅胶柱层析分离,得到粗产物。在本发明中,所述反相硅胶柱层析分离的洗脱方式为梯度洗脱,所述梯度洗脱采用的洗脱剂为水
‑
醇溶剂;所述水
‑
醇溶剂中水和醇的体积比为4~0:1。在本发明中,所述醇优选包括甲醇和/或乙醇;所述梯度洗脱过程中,水和醇的体积比依次分别为4:1、3:1、2:1、1:1和0:1,所述水和醇的体积比为1:1的组分即为所述粗产物。
[0048]
得到粗产物后,将所述粗产物进行纯化,得到具有式i所示结构的混源萜类化合物。在本发明中,所述纯化包括依次进行的凝胶柱层析和薄层层析,或,高效液相色谱纯化。在本发明中,所述凝胶柱层析采用的洗脱剂为二氯甲烷
‑
醇溶剂;所述二氯甲烷
‑
醇溶剂中二氯甲烷和醇的体积比为2~0:1;所述醇优选包括甲醇和/或乙醇。在本发明中,所述薄层层析采用的展开剂为石油醚
‑
乙酸乙酯混合溶剂,所述石油醚
‑
乙酸乙酯混合溶剂中石油醚和乙酸乙酯的体积比为2:1,所述薄层层析分离的r
f
值为0.6~0.7的组分即为具有式i所示结构的混源萜类化合物。在本发明中,所述高效液相色谱纯化的流动相为水
‑
甲醇混合溶剂,所述水
‑
甲醇混合溶剂中甲醇和水的体积比为1:1,所述高效液相色谱纯化的保留时间为18.5min的组分为具有式i所示结构的混源萜类化合物。
[0049]
本发明还提供了上述技术方案所述的混源萜类化合物或上述技术方案所述提取方法得到的混源萜类化合物在制备新型抗水产病害细菌药物中的应用。在本发明中,所述抗水产病害细菌优选包括迟缓爱德华氏菌和/或哈氏弧菌。
[0050]
下面将结合本发明中的实施例,对本发明中的技术方案进行清楚、完整地描述。显然,所描述的实施例仅仅是本发明一部分实施例,而不是全部的实施例。基于本发明中的实施例,本领域普通技术人员在没有做出创造性劳动前提下所获得的所有其他实施例,都属于本发明保护的范围。
[0051]
实施例1
[0052]
将海水样品用无菌蒸馏水稀释10倍后涂抹到pda培养基上,然后置于真菌培养箱中、28℃条件下静置培养,待所述消毒后的pda培养基表面长出真菌菌落后,将各真菌菌落分别转移至消毒后的pda培养基上继续培养,重复转移直至所述pda培养基上的菌落为单一菌株的菌落;所述海洋真菌杂色曲霉m
‑7‑
sw9在pda培养基上形成的菌落为白色气生菌丝,生长的孢子为灰绿色孢子;采用dp305试剂盒(tiangen生物技术公司)进行菌种鉴定,通过提取并克隆菌种核糖体rdna基因转录间隔序列(its1
‑
5.8s
‑
its2全长序列),然后进行基因测序,得到菌种序列后在genbank中与已知菌株的基因序列进行比较,所述菌株的基因序列如seq id no.1所示,所述菌株的dna序列与真菌杂色曲霉aspergillus versicolor(accessionno.mh911415.1)具有99%的同源性,因此,将所述的菌株命名为海洋真菌杂色
曲霉(aspergillus versicolor)m
‑7‑
sw9。
[0053]
实施例2
[0054]
取生长于平板培养基中的海洋真菌杂色曲霉(aspergillus versicolor)m
‑7‑
sw9(大小1.5cm
×
1.5cm)接种于250ml消毒后的pdb液体培养基中,于28℃、摇床(200r/min)上发酵7天,将所得菌液接入到300l消毒后的pdb液体培养基中,使用500l发酵罐发酵7天,利用乙醇浸泡发酵体系进行提取,然后用乙酸乙酯萃取4次,合并有机相后进行浓缩,得到发酵粗提物;其中,pdb液体培养基以水为溶剂,组成为:土豆粉200g、葡萄糖20g/l、蛋白胨5g/l、酵母浸粉3g/l和氯化钠35g/l。
[0055]
将所述粗提物进行正相硅胶柱层析,采用体积比依次为50~0:1的石油醚
‑
乙酸乙酯溶剂、体积比依次为50:1、40:1、30:1、20:1、10:1和0:1的二氯甲烷
‑
甲醇溶剂依次进行梯度洗脱,收集二氯甲烷和甲醇体积比为10:1的组分,得到洗脱组分;
[0056]
采用水和甲醇体积比依次为4:1、3:1、2:1、1:1和0:1水
‑
甲醇溶剂对所述洗脱组分进行反相硅胶柱层析,收集水和甲醇体积比1:1的组分,然后以体积比为1:1的水
‑
甲醇混合溶剂为流动相,收集210nm波长下保留时间为18.5min的组分,得到具有式i所示结构的混源萜类化合物。
[0057]
所述混源萜类化合物的理化和波谱特性:无色晶体;熔点为178~181℃;比旋光度[α]
20d
–
41(c 0.10,meoh);核磁共振氢谱(溶剂为氘代二甲亚砜)δ
h 6.14(s),5.38(s),5.08(s),4.51(s),3.73(s),2.01(s),1.96(d,13.9),1.84(s),1.77(d,10.9),1.68(t,13.6),1.62(m),1.53(m),1.50(m),1.46(m),1.40(m),7.53(d,7.6),1.21(br s),1.17(s),0.98(s),0.87(s),0.80(s);核磁共振碳谱(溶剂为氘代二甲亚砜)δ
c 170.6(c),170.3(c),166.8(c),137.7(c),136.3(c),127.2(ch),116.3(ch2),89.2(c),77.0(ch),69.8(c),52.4(ch3),48.7(c),38.4(c),36.5(c),32.8(ch2),30.4(c),28.0(ch3),25.5(ch2),22.0(ch2),21.8(ch3),21.5(ch3),18.4(ch3),17.4(ch3),17.2(ch2),16.0(ch3);高分辨质谱m/z 431.2423[m+h]
+
,c
25
h
35
o
6+
计算值为435.2428。
[0058]
对比例1
[0059]
化合物1和化合物2,其结构式如下:
[0060][0061]
测试例1
[0062]
水产病害细菌抑制剂活性
[0063]
用最小抑菌浓度法检测式i所示化合物的抗水产病害细菌活性。选择以下4株水产病原菌株:迟缓爱德华氏菌、哈氏弧菌、藤黄微球菌和副溶血性弧菌进行抗菌活性测试。
[0064]
(1)抗菌活性测试(mic法):最小抑菌浓度即体外能够抑制细菌生长的最低药物浓度。在96微孔板中,通过将不同浓度的药物加入到待测菌的菌悬液中,培养后观察,如果指示菌在某孔内生长,表示该孔的药物浓度不能抑制该菌的生长,该孔内液体浑浊,透光度明
显下降。反之,该孔内液体澄清,透光度下降不显著。小孔内完全抑制指示菌生长的最低样品浓度为该化合物的mic值。
[0065]
(2)菌悬液的制备
[0066]
分别将迟缓爱德华氏菌和哈氏弧菌接种于培养基(迟缓爱德华氏菌用tsb培养基;哈氏弧菌采用lb培养基)上于28℃培养24h后,吸取4ml无菌的0.85%nacl溶液洗涤培养物,并用玻璃刮刀将菌轻轻刮下。用移液枪吸取适量菌悬液于无菌试管中,然后用0.85%nacl溶液将菌悬液调至0.5麦氏浊度(即1.5
×
108cfu/ml),并进一步用0.85%nacl溶液稀释至5
×
105cfu/ml;
[0067]
其中,0.85%氯化钠溶液的配制方法:将8.5g氯化钠溶解于水中后定容到1000ml。
[0068]
0.5麦氏浊度标准:将0.5ml浓度为0.048mol/l的bacl2加到99.5ml浓度为0.18mol/l的h2so4中并不断搅动以维持混悬状态。
[0069]
(3)待测药品溶液的配制
[0070]
待测药品:具有式i所示结构的混源萜类化合物、阳性对照药(氯霉素)、化合物1和化合物2。
[0071]
分别将混源萜类化合物和氯霉素溶解于100μldmso中,充分混匀后,得到待测药品溶液,其中,待测药品溶液的最终浓度为2560μg/ml;吸取50μl待测药品溶液置于离心管中,加入50μl dmso,得到浓度减半的待测药品溶液,重复上述操作,得到11组浓度依次减半的待测药品溶液(浓度依次为2560μg/ml、1280μg/ml、640μg/ml、320μg/ml、160μg/ml、80μg/ml、40μg/ml、20μg/ml、10μg/ml、5μg/ml和2.5μg/ml)。
[0072]
(4)空白对照:以dmso作为空白对照。
[0073]
(5)mic值测定流程:
[0074]
(5.1)采用无菌操作,将不同浓度的待测药品溶液分别加到无菌的96孔板中,第1~11孔加待测药品溶液,每孔5μl,第12孔不加待测药品溶液作为生长对照。
[0075]
(5.2)将相当于0.5麦氏比浊度的指示菌悬液,经液体培养基(迟缓爱德华氏菌采用tsb培养基;哈氏弧菌采用lb培养基)稀释1000倍后,取95μl依次加入到96孔板中,使得第1~11孔的待测药品溶液终浓度依次为128μg/ml、64μg/ml、32μg/ml、16μg/ml、8μg/ml、4μg/ml、2μg/ml、1μg/ml、0.5μg/ml、0.25μg/ml和0.125μg/ml。轻轻震荡混匀后,将96孔板密封置于28℃培养箱中细菌培养24h。
[0076]
(5.3)在600nm波长下使用酶标仪测定每孔的吸光值,以在小孔内完全抑制指示菌生长的最低样品浓度为待测药品的mic值。当阴性对照孔内指示菌明显生长实验才有意义;当实验出现单一的跳孔时,应记录抑制菌株生长的最高药物浓度;如出现多处跳孔,则不报告结果,需重复实验。
[0077]
实验结果:混源萜化合物分别对迟缓爱德华氏菌和哈氏弧菌具有较强的抑制活性,mic值均为1.0μg/ml,氯霉素对迟缓爱德华氏菌的mic值为2.3μg/ml,对哈氏弧菌的mic值为2.2μg/ml,化合物1和化合物2对于迟缓爱德华氏菌和哈氏弧菌的mic值均为8.0μg/ml。说明,本发明提供的混源萜化合物对迟缓爱德华氏菌和哈氏弧菌的抑制效果优于氯霉素、化合物1和化合物2。
[0078]
以上所述仅是本发明的优选实施方式,应当指出,对于本技术领域的普通技术人员来说,在不脱离本发明原理的前提下,还可以做出若干改进和润饰,这些改进和润饰也应
视为本发明的保护范围。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1