一种磷光化合物及其制备方法、有机电致发光器件与流程
1.本发明属于有机电致发光材料技术领域,尤其涉及一种磷光化合物及其制备方法、有机电致发光器件。
背景技术:
2.有机电子器件以有机发光器件为典型代表,而有机发光器件中的典型实例是有机发光二极管(organic light emitting diode,简称oled)。有机发光二极管是一种以有机材料作为活性材料的电流驱动式发光器件,具体是指有机半导体材料和有机发光材料在电场的驱动下,通过载流子注入和复合导致发光的技术oled显示技术的优势十分明显,如器件厚度薄、结构简单不需要背光源、能主动发光,此外还有功耗低、视角宽、响应速度快等优点,被视为下一代显示技术的有力竞争者。
3.oled发光分为荧光发光和磷光发光两种方式,根据理论推测,由电荷的再结合而引起的单重激发态与三重激发态的比例为1:3,所以使用小分子荧光材料时,能用于发光的仅为全部能量的25%,其余的75%的能量因三重激发态的非发光机制而损失掉,故一般认为荧光材料的内部量子效率极限为25%。在磷光发光中,利用了单线态和三线态激子,相对荧光材料只利用单线态激子,比例高达75%的三线态激子的有效利用,使得基于磷光材料的pholed理论上实现了100%的内量子效率。近三年来,磷光材料逐渐取代传统的荧光材料,成为oled发光材料的研究热点。
4.可以通过将主体材料与掺杂剂组合来制备发光材料以改进色彩纯度、发光效率以及稳定性。主体材料极大地影响有机发光器件的效率和性能,开发一种符合实用性要求的新型主体材料是至关重要的。但是由于磷光材料合成工艺比较复杂、耗时久、寿命低,所以磷光材料的进一步开发迫在眉睫。因此,本技术提供一种磷光化合物及其制备方法、有机电致发光器件。
技术实现要素:
5.本发明实施例的目的在于提供一种磷光化合物及其制备方法、有机电致发光器件,旨在解决背景技术中指出的现有技术存在的问题。
6.本发明实施例是这样实现的,一种磷光化合物,所述磷光化合物的结构通式为通式i:
[0007][0008]
其中,r1、r2、r3、r4各自独立地为氢、c1
‑
c10烷基、氰基、胺基、取代或未取代的c6
‑
c30芳基或取代或未取代的c3
‑
c30杂芳基;
[0009]
x为化学键、o、s、cr5r6或nr7;
[0010]
r5、r6各自独立地为取代或未取代的c1
‑
c6烷基、取代或未取代的c6
‑
c30芳基,或取代或未取代的c3
‑
c30杂芳基;
[0011]
r、r7各自独立地为c6
‑
c30亚芳基或c3
‑
c20亚杂芳基;
[0012]
r1‑
r4取代位置为所在苯环上的任意位置,取代个数为0
‑
4的整数。
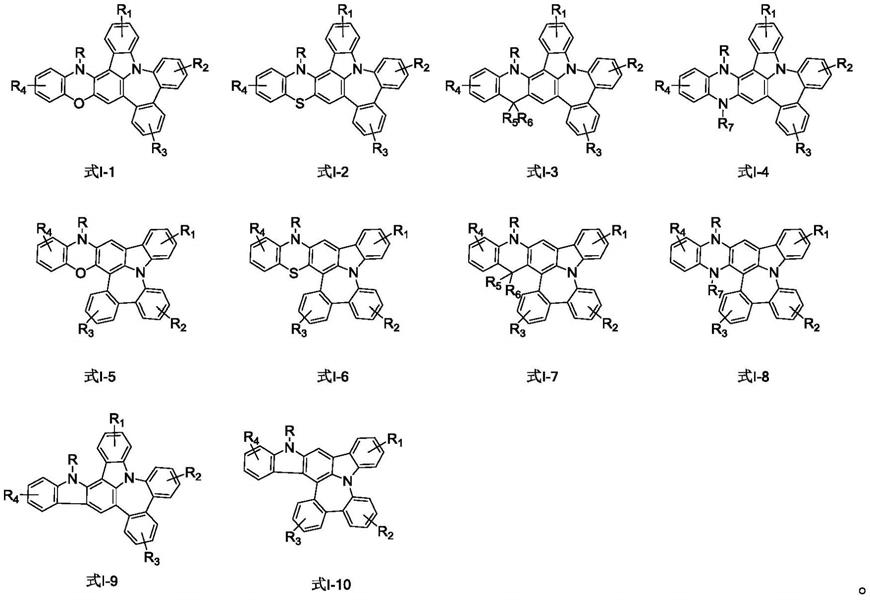
[0013]
作为本发明实施例的另一种优选方案,所述磷光化合物的结构通式为通式i
‑
1~通式i
‑
10:
[0014][0015]
作为本发明实施例的另一种优选方案,所述r1、r2、r3、r4各自独立地为氢、甲基、乙基、氰基、苯基、萘基、联苯基、三联苯基、菲基、蒽基、芘基、苝基、三亚苯基、荧蒽基、三嗪基、嘧啶基、吡啶基、喹唑啉基、喹啉基、喹喔啉基或胺基;
[0016]
r1、r2、r3或r4可以与其相邻的芳香环稠合成环或取代。
[0017]
作为本发明实施例的另一种优选方案,所述r5、r6各自独立地为甲基、乙基、丙基、
环己基、苯基、萘基、联苯基、二苯并呋喃基、二苯并噻吩基或咔唑基;r7为环己基、苯基、萘基、联苯基、甲基苯或三联苯。
[0018]
作为本发明实施例的另一种优选方案,所述r为苯基、萘基、联苯基、三联苯基、菲基、蒽基、芘基、苝基、三亚苯基、荧蒽基、三嗪基、嘧啶基、吡啶基、喹唑啉基、喹啉基或喹喔啉基以及如下基团中的任一种:
[0019]
[0020]
[0021][0022]
作为本发明实施例的另一种优选方案,所述磷光化合物的化学结构式为式1~式156中的一种:
[0023]
[0024]
[0025]
[0026]
[0027]
[0028]
[0029][0030]
本发明实施例的另一目的在于提供一种所述的磷光化合物的制备方法,其合成路线为:
[0031][0032]
具体包括以下步骤:
[0033]
在保护气氛下,将反应物a
‑
i、反应物b
‑
i、四(三苯基膦钯)和碳酸钾置于溶剂中进
行反应;反应结束后,所得物经纯化得到中间体c
‑
i;
[0034]
在保护气氛下,加入中间体c
‑
i、pd(oac)2、pcy3和碳酸钾,置于溶剂中进行反应;反应完成后,混合物经洗涤、萃取,所得有机层经洗脱、纯化得到结构通式为通式i的磷光化合物。
[0035]
优选的,其具体包括以下步骤:
[0036]
(1)反应瓶中,在氮气保护下,加入反应物a
‑
i(1.0eq)、反应物b
‑
i(2.2eq)、四(三苯基膦钯)(0.01eq)、碳酸钾(2.5eq),以及溶剂:甲苯/乙醇/水(体积比3:2:2),置于80℃温度下反应8h;反应结束后,冷却至室温,过滤,所得到的固体通过甲苯重结晶纯化,得到中间体c
‑
i;
[0037]
(2)反应瓶中,在氮气保护下,加入中间体c
‑
i(1.0eq)、pd(oac)2(0.01eq)、pcy3(0.02eq)、碳酸钾(3.0eq)和溶剂dmf,置于150℃温度下反应12h;反应完成后,混合物用蒸馏水洗涤并且用乙酸乙酯萃取;用硫酸镁干燥所得有机层,并且通过旋转式蒸发器从其中去除溶剂,以二氯甲烷:石油醚体积比为1:(1
‑
9)作为洗脱剂,用层析柱纯化得到结构通式为通式i的磷光化合物。
[0038]
本发明实施例的另一目的在于提供一种如上述的方法制备得到的磷光化合物。
[0039]
本发明实施例的另一目的在于提供一种有机电致发光器件,包括阳极、阴极以及至少一层设置在所述阳极和所述阴极之间的有机物层,所述的有机物层包括发光层,所述发光层包括主体材料和掺杂材料,所述的磷光化合物作为主体材料。有机电致发光器件通过在基板上依次层叠阳极、有机物层和阴极制造而成,可以采用以下现有方法制造:溅射法(sputtering)或电子束蒸发法(ebeam evaporation)之类的物理蒸镀方法(physical vapor deposition,pvd)。
[0040]
作为本发明实施例的另一种优选方案,所述阳极、阴极之间设置有空穴注入层、空穴传输层、电子阻挡层、发光辅助层、发光层、空穴阻挡层、电子传输层、电子注入层中的至少一层或多层。
[0041]
其中,阳极优选包含具有高逸出功的材料,例如氧化锡铟(ito)或氧化铟锌(izo)。
[0042]
空穴传输材料是能够接收来自阳极或空穴注入层的空穴并将空穴传输至发光层的材料,并且具有高空穴迁移率的材料。
[0043]
电子阻挡层可以设置在空穴传输层与发光层之间。
[0044]
发光层的材料是一种通过分别接收来自空穴传输层和电子传输层的空穴和电子,并将所接收的空穴和电子结合而能发出可见光的材料。
[0045]
优选的,发光层包含主体材料和掺杂材料;主体材料和掺杂材料的质量比为90
‑
99.5:0.5
‑
10;
[0046]
优选的,主体材料分为磷光主体材料和荧光主体材料,磷光化合物优选作为磷光主体材料。
[0047]
掺杂材料可以包括荧光掺杂和磷光掺杂。
[0048]
所述磷光掺杂材料包括铱、铂等的金属络合物的磷光材料。例如,可以使用ir(ppy)3等绿色磷光材料,firpic、fir6等蓝色磷光材料和btp2ir(acac)等红色磷光材料。
[0049]
空穴阻挡层材料,可以使用现有技术中公知的具有空穴阻挡作用的化合物,例如,浴铜灵(bcp)等菲咯啉衍生物、噁唑衍生物、三唑衍生物、三嗪衍生物等,但不限于此。
[0050]
电子传输层可以起到促进电子传输的作用。可以使用现有技术中公知的具有电子传输作用的化合物,例如,8
‑
羟基喹啉的al配合物;包含alq3的配合物;有机自由基化合物;羟基黄酮
‑
金属配合物等。
[0051]
电子注入层可以起到促进电子注入的作用。具有传输电子的能力,防止发光层中产生的激子迁移至空穴注入层。
[0052]
阴极优选具有小功函数的材料,使得电子顺利注入有机材料层。例如镁、钙、钠、钾、钛、铟、钇、锂、钆、铝、银、锡和铅,或其合金。
[0053]
与现有技术相比,本发明实施例的有益效果是:
[0054]
本发明实施例提供了一种新的磷光化合物,可用作为有机电致发光器件的主体材料,相比于现有磷光材料,采用本发明的磷光化合物作为主体材料制备的有机电致发光器件,其驱动电压、发光效率、寿命以及玻璃态转变温度各方面性能均得到改善,从而可以提高有机电致发光器件的实用性。另外,本发明实施例提供的磷光化合物的制备方法,合成过程简单,产品产率较高。
具体实施方式
[0055]
为了使本发明的目的、技术方案及优点更加清楚明白,以下结合实施例,对本发明进行进一步详细说明。应当理解,此处所描述的具体实施例仅仅用以解释本发明,并不用于限定本发明。
[0056]
以下结合具体实施例对本发明的具体实现进行详细描述。
[0057]
实施例1磷光化合物1的合成
[0058]
该实施例提供了一种磷光化合物,其制备方法如下:
[0059][0060]
(1)反应瓶中,在氮气保护下,加入反应物a
‑
1(100mmol),反应物b
‑
1(220mmol),四(三苯基膦钯)(1mmol),甲苯:乙醇:水=600ml:400ml:400ml,碳酸钾(250mmol),置于80℃
温度下反应8h;反应结束后,冷却至室温,过滤,所得到的固体通过甲苯重结晶纯化,得到中间体c
‑
1(56.5g,ms:689.26,产率:82%);
[0061]
(2)反应瓶中,在氮气保护下,加入中间体c
‑
1(70mmol),pd(oac)2(0.07mmol),pcy3(1.4mmol),碳酸钾(210mmol),dmf(700ml),置于150℃温度下反应12h;反应完成后,混合物用蒸馏水洗涤并且用乙酸乙酯萃取;然后用硫酸镁干燥所得有机层,并且通过旋转式蒸发器从其中去除溶剂,以二氯甲烷:石油醚体积比为1:(1
‑
9)作为洗脱剂,用层析柱纯化得到磷光化合物1(39.3g,产率:86%)。
[0062]
对所得磷光化合物1进行检测分析,结果如下:
[0063]
质谱测试:理论值为652.80;测试值为652.68。
[0064]
元素分析(单位:%):
[0065]
理论值:c,86.48;h,4.94;n,8.58;
[0066]
测试值:c,86.49;h,4.93;n,8.58。
[0067]
实施例2磷光化合物52的合成
[0068][0069]
(1)反应瓶中,在氮气保护下,加入反应物a
‑
52(100mmol)、反应物b
‑
52(220mmol),四(三苯基膦钯)(1mmol),甲苯:乙醇:水=600ml:400ml:400ml,碳酸钾(250mmol),置于80℃温度下反应8h;反应结束后,冷却至室温,过滤,所得到的固体通过甲苯重结晶纯化,得到中间体c
‑
52(73.2g,ms:915.31,产率:80%);
[0070]
(2)反应瓶中,在氮气保护下,加入中间体c
‑
52(70mmol),pd(oac)2(0.07mmol),
pcy3(1.4mmol),碳酸钾(210mmol),dmf(700ml),置于150℃温度下反应12h;反应完成后,混合物用蒸馏水洗涤并且用乙酸乙酯萃取;然后用硫酸镁干燥所得有机层,并且通过旋转式蒸发器从其中去除溶剂,以二氯甲烷:石油醚体积比为1:(1
‑
9)作为洗脱剂,用层析柱纯化得到磷光化合物52(51.8g,产率:84%)。
[0071]
对所得化合物52进行检测分析,结果如下:
[0072]
质谱测试:理论值为880.07;测试值为880.42。
[0073]
元素分析(单位:%):
[0074]
理论值:c,87.35;h,4.70;n,7.96;
[0075]
测试值:c,87.33;h,4.70;n,7.94。
[0076]
实施例3磷光化合物100的合成
[0077][0078]
(1)反应瓶中,在氮气保护下,加入反应物a
‑
100(100mmol),反应物b
‑
100(220mmol),四(三苯基膦钯)(1mmol),甲苯:乙醇:水=600ml:400ml:400ml,碳酸钾(250mmol),置于80℃温度下反应8h;反应结束后,冷却至室温,过滤,所得到的固体通过甲苯重结晶纯化,得到中间体c
‑
100(49.0g,ms:628.15,产率:78%);
[0079]
(2)反应瓶中,在氮气保护下,加入中间体c
‑
100(70mmol),pd(oac)2(0.07mmol),pcy3(1.4mmol),碳酸钾(210mmol),dmf(700ml),置于150℃温度下反应12h;反应完成后,混合物用蒸馏水洗涤并且用乙酸乙酯萃取;然后用硫酸镁干燥所得有机层,并且通过旋转式蒸发器从其中去除溶剂,以二氯甲烷:石油醚体积比为1:(1
‑
9)作为洗脱剂,用层析柱纯化得到磷光化合物100(33.2g,产率:80%)。
[0080]
对所得磷光化合物100进行检测分析,结果如下:
[0081]
质谱测试:理论值为592.72;测试值为592.54。
[0082]
元素分析(单位:%):
[0083]
理论值:c,81.06;h,4.08;n,9.45;s,5.41
[0084]
测试值:c,81.07;h,4.08;n,9.44;s,5.40。
[0085]
实施例4磷光化合物121的合成
[0086][0087]
(1)反应瓶中,在氮气保护下,加入反应物a
‑
121(100mmol),反应物b
‑
121(220mmol),四(三苯基膦钯)(1mmol),甲苯:乙醇:水=600ml:400ml:400ml,碳酸钾(250mmol),置于80℃温度下反应8h;反应结束后,冷却至室温,过滤,所得到的固体通过甲苯重结晶纯化,得到中间体c
‑
121(62.6g,ms:813.27,产率:77%);
[0088]
(2)反应瓶中,在氮气保护下,加入中间体c
‑
121(70mmol),pd(oac)2(0.07mmol),pcy3(1.4mmol),碳酸钾(210mmol),dmf(700ml),置于150℃温度下反应12h;反应完成后,混合物用蒸馏水洗涤并且用乙酸乙酯萃取;然后用硫酸镁干燥所得有机层,并且通过旋转式蒸发器从其中去除溶剂,以二氯甲烷:石油醚体积比为1:(1
‑
9)作为洗脱剂,用层析柱纯化得到磷光化合物121(44.1g,产率:81%)。
[0089]
对所得磷光化合物121进行检测分析,结果如下:
[0090]
质谱测试:理论值为777.29;测试值为777.21。
[0091]
元素分析(单位:%):
[0092]
理论值:c,86.46;h,4.54;n,9.00;
[0093]
测试值:c,86.45;h,4.55;n,9.01。
[0094]
实施例5~实施例30
[0095]
因结构通式为发明内容中的通式i,其他磷光化合物的制备方法的合成路线和原理均与上述所列举的实施例1相同,只需要将原料分别替换为目标产物对应的反应物,反应
物用量按照相应化学计量比相应调整即可得到相对应的磷光化合物,所以在此不再穷举,本发明实施例参照实施例1至4的制备方法完成对磷光化合物4、8、13、18、23、30、36、44、50、56、62、70、76、82、89、95、99、106、110、116、120、124、130、136、144和150的合成,其质谱、分子式、产率如表1所示。
[0096]
表1
[0097]
实施例磷光化合物分子式质谱理论值质谱测试值产率%实施例54c
44
h
26
n4610.72610.3478实施例68c
45
h
28
n4624.75624.8179实施例713c
45
h
27
n3609.73609.5786实施例818c
48
h
28
n4660.78660.5882实施例923c
49
h
29
n5687.81687.8484实施例1030c
57
h
33
n5o803.93803.3386实施例1136c
52
h
32
n4712.86712.6286实施例1244c
37
h
21
n3507.60507.4182实施例1350c
65
h
39
n7918.08918.4782实施例1456c
53
h
33
n3711.87711.4785实施例1562c
44
h
26
n4610.72610.7579实施例1670c
62
h
37
n5o868.01868.7885实施例1776c
57
h
33
n5os835.99835.4580实施例1882c
45
h
28
n4s656.81642.7481实施例1989c
61
h
37
n7s900.08900.0780实施例2095c
61
h
37
n5s872.06872.1480实施例2199c
41
h
25
n3s591.73591.4784实施例22106c
44
h
26
n4s642.78642.6584实施例23110c
68
h
43
n5o946.13896.4884实施例24116c
63
h
38
n6s911.10911.2078实施例25120c
52
h
31
n7753.87753.5186实施例26124c
58
h
37
n5803.97803.6581实施例27130c
47
h
32
n4652.80652.4782实施例28136c
61
h
38
n6s887.08887.5384实施例29144c
49
h
32
n4s708.88708.6280实施例30150c
50
h
28
n6712.82712.8081
[0098]
另外,需要说明,本技术其他磷光化合物参照上述所列举的实施例的制备方法即可获得,所以在此不再一一列举。
[0099]
器件实施例1:
[0100]
该器件实施例提供了一种有机电致发光器件,其具体的制备方法包括以下步骤:
[0101]
(1)将用于oled装置玻璃基板上的ito/ag/ito薄膜(ito厚度为14nm,ag厚度为150nm)放在蒸馏水中清洗2次,每次用超声波洗涤30分钟;然后用蒸馏水反复清洗2次,每次用超声波洗涤10分钟;蒸馏水清洗结束后,按顺序分别在异丙醇、丙酮、甲醇等溶剂中用超
声波洗涤后干燥;然后用等离子体清洗机洗涤5分钟,再送到蒸镀机里。
[0102]
(2)将化合物ht以及p
‑
dopant(3%)引入真空气相沉积设备的小室中,并且然后将所述设备的腔室中的压力控制到10
‑6托。此后,向小室施加电流以使以上引入的材料蒸发,从而在ito基板上形成具有10nm厚度的空穴注入层。接下来,将化合物ht引入真空气相沉积设备的另一个小室中,并通过向小室施加电流使化合物蒸发,从而在空穴注入层上形成具有130nm厚度的空穴传输层。
[0103]
(3)然后将化合物ebl引入真空气相沉积设备的小室中,并通过向小室施加电流使所述化合物蒸发,从而在空穴传输层上形成具有10nm厚度的电子阻挡层。将实施例1提供的磷光化合物1引入真空气相沉积设备的一个小室中作为磷光主体,并将化合物dopant引入另一个小室中作为磷光掺杂材料。磷光主体材料和磷光掺杂材料的掺杂比为97:3,在电子阻挡层上形成具有20nm厚度的发光层。
[0104]
(4)在上述发光层上同时真空蒸镀et和liq作为电子传输层(35nm,50%);在上述电子传输层上真空蒸镀厚度为1.0nm的yb,作为电子注入层。在电子注入层上真空蒸镀镁和银作为阴极,镁和银的重量比为1:9,蒸镀厚度为18nm,在上述阴极上真空蒸镀cpl层,蒸镀厚度为70nm,制得有机电致发光器件。
[0105]
制得的有机电致发光器件的结构为:
[0106]
ito/ag/ito/ht:p
‑
dopant(10nm,3%)/ht(130nm)/ebl(10nm)/化合物1:dopant(20nm,2%)/et:liq(35nm,50%)/yb(1nm)/mg:ag(18nm,1:9)/cpl(70nm)。
[0107]
其中,上述部分原料的化学结构式如下所示:
[0108][0109]
器件实施例2~器件实施例156
[0110]
参照上述方法,将器件实施例1中使用的化合物1分别替换为化合物2
‑
156作为电子传输层,制备得到相应的有机电致发光器件。
[0111]
器件对比例1~器件对比例4
[0112]
参照上述方法,将器件实施例1中使用的化合物1分别替换为对比化合物1
‑
4,制备
得到相应的有机电致发光器件。
[0113][0114][0115]
对上述制备的有机电致发光器件加以正向直流偏置电压,利用photo research公司的pr
‑
650光度测量设备测定有机电致发光特性,在亮度6000cd/m2条件下利用mcscience公司的寿命测定装置测定t95的寿命。结果如表2所示。
[0116]
表2
[0117]
[0118]
[0119]
[0120][0121][0122]
从表2可以看出,采用本发明的磷光化合物作为主体材料制备的有机电致发光器件与采用对比化合物1~4制备的有机电致发光器件相比,其驱动电压、发光效率、寿命以及
玻璃态转变温度各方面性能均得到改善。
[0123]
以上所述仅为本发明的较佳实施例而已,并不用以限制本发明,凡在本发明的精神和原则之内所作的任何修改、等同替换和改进等,均应包含在本发明的保护范围之内。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1