基于异恶唑取代苯甲酰胺类衍生物及抗前列腺癌药物应用
1.本发明涉及药物领域,具体涉及一种基于异恶唑取代苯甲酰胺类衍生物及其制备方法及抗前列腺癌药物应用。
背景技术:
2.前列腺癌(prostate cancer)是威胁男性健康的常见肿瘤之一,为欧美等发达国家男性中得病率最高致死率第二的恶性肿瘤。雄激素剥夺疗法(adt)为目前临床上广泛应用的前列腺癌治疗方案。在经过平均时间为18
‑
24个月的缓解期后,原来对内分泌治疗敏感的前列腺癌,转为去势抵抗前列腺癌(crpc),传统雄激素剥夺疗法不再有效,因而针对crpc机制及治疗研究已成为前列腺癌研究中的重点及难点。
3.雄激素受体(ar)是一类核受体,包含四个主要的结构域:氮端结构域(ntd),dna结合结构域(dbd),柔性铰链区(hinge),以及配体结合结构域(lbd)。ar对前列腺癌的发生和发展非常重要,同时其信号通路在多数crpc病人中保持活跃,抑制ar信号通路对于治疗crpc仍有重要的意义。目前临床应用于crpc治疗的药物主要为第二代ar拮抗剂,包括abiraterone、enzalutamide以及apalutamide等。这些药物的成功研发促进了crpc患者的治疗,并进一步明确了抑制雄激素信号通路对于治疗crpc的关键作用。
4.临床治疗及基础研究表明crpc对enzalutamide和apalutamide均产生耐药性。其一:ar激素结合口袋(hbp)处残基f876l突变能够使crpc对enzalutamide和apalutamide产生耐药性,使其由拮抗剂变成激动剂。其二:缺乏配体结合结构域的arvs具有不依赖雄激素的转录活性,因而作用于hbp位点的enzalutamide、apalutamide等对arvs转录活性无法抑制。此外,由于abiraterone通过抑制雄激素合成途经发挥作用,ar可变剪接体的产生也是abiraterone产生耐药的关键因素。其三:激活传统的nf
‑
κb2信号通路,会使雄激素敏感型前列腺癌细胞转变成雄激素不敏感型,从而对抗雄激素药物产生耐药性。其四:gr(glucocorticoid receptor)高表达作为ar信号通路的旁路来发挥作用是crpc对enzalutamide产生耐药的新机制。
5.近年来,以蛋白水解靶向嵌合分子(protacs)为主的蛋白诱导降解小分子技术取得了巨大的突破,成为药物研发的新策略。和传统药物通过占据结合位点需要大过量的药物分子相比,蛋白诱导降解小分子仅需催化量就可以起到很好的抑制作用,展现出常规小分子抑制剂不具备的独特优势。该策略被应用于ar拮抗剂研发,并取得突破性的进展,这类分子可以选择性的降解ar,被称为选择性雄激素受体降解分子(sards)。但作为新技术,protacs仍有缺陷,尤其是在药物开发方面。首先,双功能分子分子量都较高,因而类药性不能保证,导致其水溶性、口服吸收和透膜性都较差,药动和药代不佳。其次,双功能分子大分子量以及化学结构的复杂性也会导致化学合成难度和成本增加。此外,由于目前报道双功能分子sards的连接分子均作用于ar的lbd结构域,因而并不能降解缺乏lbd结构域的ar可变剪接体ar
‑
v7等,从而仍无法克服由于表达ar可变剪接体引起的耐药问题。
技术实现要素:
6.本发明的目的提供一类基于异恶唑取代苯甲酰胺类衍生物及抗前列腺癌药物应用,发现及优化异恶唑取代苯甲酰胺类衍生物雄激素受体降解分及其抗前列腺癌应用作为新一代的雄激素拮抗剂。
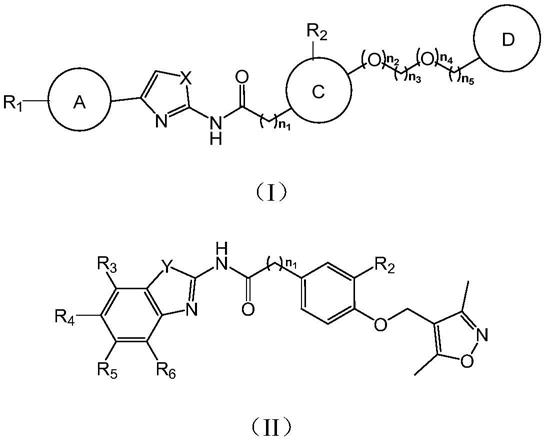
7.本发明中所述的异恶唑取代苯甲酰胺类衍生物的结构如式(i)、结构式(ii)所示:
[0008][0009]
a环独立选自:苯、噻吩、呋喃、苯并噻吩、苯并呋喃;
[0010]
c环独立选自:苯、吡啶、联苯、萘;
[0011]
d环独立选自:吡唑、吲哚、吲哚啉、吗啉、顺式
‑
2,6
‑
二甲基吗啉;
[0012]
x独立选自:s原子、n原子;
[0013]
y独立选自:s原子、o原子;
[0014]
r1独立选自:
‑
f、
‑
och3、
‑
ocf3、
‑
br、
‑
cl、
‑
cf3、
‑
ch3、
‑
cn;
[0015]
r2独立选自:
‑
och3、
‑
ch3、
‑
f、
‑
ocf3、
‑
cf3、
‑
cl、
‑
no2、
‑
cn、
‑
cho、
‑
ch2oh;
[0016]
r3独立选自:h;
[0017]
r4独立选自:h、
‑
ch3、
‑
och3、
‑
ocf3、
‑
f、
‑
cl;
[0018]
r5独立选自:h、
‑
f、
‑
och3、
‑
ch3;
[0019]
r6独立选自:h、
‑
ch3;
[0020]
n1独立选自:0
‑
1;
[0021]
n2独立选自:0
‑
1;
[0022]
n3独立选自:2
‑
5;
[0023]
n4独自选自:0
‑
1;
[0024]
n5独立选自:1
‑
3。
[0025]
本发明中所述的衍生物a环修饰的苯甲酰胺类衍生物结构如式(iii)所示:
[0026][0027]
a环为5
‑
10个原子的苯环或苯环取代物及杂环或杂环取代物,具体结构如图1所示。苯丙酰胺类衍生物a环结构如图1所示。a环修饰的苯甲酰胺类衍生物合成路线如图2所示。
[0028]
本发明中所述的衍生物b环修饰的苯甲酰胺类衍生物结构如式(iv)所示:
[0029][0030]
x为氮原子,b环为5个原子的杂环咪唑,合成路线如图3所示。b环修饰的苯甲酰胺类衍生物合成路线如图3所示。
[0031]
本发明中所述的衍生物e(ab环合并)环修饰的苯甲酰胺类衍生物结构如式(v)所示:
[0032][0033]
r3独立选自:h;
[0034]
r4独立选自:h、
‑
ch3、
‑
och3、
‑
ocf3、
‑
f、
‑
cl;
[0035]
r5独立选自:h、
‑
f、
‑
och3、
‑
ch3;
[0036]
r6独立选自:h、
‑
ch3;
[0037]
y独立选自:s、o。
[0038]
e环修饰的苯甲酰胺类衍生物合成路线如图4所示。
[0039]
本发明中所述的衍生物c环修饰的苯甲酰胺类衍生物结构如式(vi)和式(vii)所示:
[0040][0041]
r独立选自:
‑
och3、
‑
ch3、
‑
f、
‑
ocf3、
‑
cf3、
‑
cl、
‑
no2、
‑
cn、
‑
cho、
‑
ch2oh;
[0042]
e独立选自:n。
[0043]
c环独立选自:联苯、萘。
[0044]
c环修饰的苯甲酰胺类衍生物合成路线如图5所示。
[0045]
本发明中所述的衍生物d环修饰的苯甲酰胺类衍生物结构如式(viii)所示。
[0046][0047][0048]
d环独立选自:吡唑、吲哚、吲哚啉、吗啉、顺式
‑
2,6
‑
二甲基吗啉;
[0049]
n2独立选自:0
‑
1;
[0050]
n3独立选自:1
‑
2。
[0051]
d环修饰的苯甲酰胺类衍生物合成路线如图6所示。
[0052]
本发明中所述的衍生物链优化的苯甲酰胺类衍生物结构如式(ix)所示
[0053][0054]
n2独立选自:0
‑
1;
[0055]
n3独立选自:0
‑
5;
[0056]
n4独立选自:0
‑
1;
[0057]
链修饰的苯甲酰胺类衍生物合成路线如图7所示。
[0058]
基于异恶唑取代苯甲酰胺类衍生物在制备雄激素受体拮抗剂和雄激素受体下调
剂中的应用。
[0059]
所述的雄激素受体拮抗剂抑制ar
‑
t877a突变型受体的活性。所述的雄激素受体拮抗剂抑制ar
‑
f876l突变型受体的活性。所述的雄激素受体拮抗剂在制备治疗雄激素失调疾病的药物中的应用。所述雄激素失调疾病为由雄激素亢奋引起的疾病。所述雄激素失调疾病为前列腺增生症或前列腺癌。所述雄激素失调疾病为男性性欲亢进。所述雄激素失调疾病为女性痤疮、女性脂溢性皮炎、女性多毛症、女性脱发。
[0060]
所述的雄激素受体下调剂促进雄激素受体蛋白的降解。
[0061]
所述的雄激素受体拮抗剂在制备治疗前列腺癌药物中的应用,所述的雄激素受体拮抗剂作为雄激素受体拮抗剂与下调剂治疗现有药物敏感型前列腺癌和耐药型前列腺癌。所述的雄激素受体拮抗剂在治疗前列腺癌复方药(和现有药物恩杂鲁胺联合用药)制备的应用。
[0062]
与现有技术相比,本发明具有如下优点:
[0063]
本发明中利用课题组构建的ar蛋白降解筛选模型、ar双荧光报告系统筛选已经合成优化,发现了一类异恶唑取代苯甲酰胺类衍生物,可以有效抑制雄激素受体的活性,同时进一步的实验证实,这类化合物可以降解全长雄激素受体以及雄激素受体可变剪接体,是一类新型的雄激素拮抗剂,可以应用于雄激素依赖的抗前列癌药物治疗。
附图说明
[0064]
图1为苯丙酰胺类衍生物a环结构;
[0065]
图2为a环修饰的苯甲酰胺类衍生物合成路线;
[0066]
图3为b环修饰的苯甲酰胺类衍生物合成路线;
[0067]
图4为e环修饰的苯甲酰胺类衍生物合成路线;
[0068]
图5为c环修饰的苯甲酰胺类衍生物合成路线;
[0069]
图6为d环修饰的苯甲酰胺类衍生物合成路线;
[0070]
图7为链修饰的苯甲酰胺类衍生物合成路线;
[0071]
图8为部分苯甲酰胺衍生物的westen
‑
blot实验结果。
具体实施方式
[0072]
以下实施例仅为帮助本领域技术人员更好地理解本发明,但不以任何方式限制本发明。
[0073]
《实施例1》4
‑
((3,5
‑
二甲基异恶唑
‑4‑
基)甲氧基)
‑
n
‑
(4
‑
苯基噻唑
‑2‑
基)苯甲酰胺的合成(za1)
[0074]
将化合物1a和对甲苯磺酸(tsoh,0.1eq)溶于无水二氯甲烷中,室温搅拌,分批次加入n
‑
溴代丁二酰亚胺(nbs,1.0eq),加热回流,12h后tlc检测反应完全,反应液由浅黄色变为棕红色。冷却至室温后加入适量饱和食盐水洗涤,二氯甲烷萃取,无水硫酸钠干燥,减压蒸发溶剂,残余物经柱层析分离得到棕红色油状物。将硫脲(1.2eq)溶于乙醇,室温搅拌,加入棕红色油状物的乙醇溶液,加热回流。2h后tlc检测反应完全,反应液由浅黄色变成深黄色。冷却后减压蒸发溶剂,加入适量饱和碳酸氢钠溶液洗涤,乙酸乙酯萃取,无水硫酸钠干燥,减压蒸发溶剂,残余物经柱层析分离得到淡黄色固体4a,收率63%。
[0075]
将3,5
‑
二甲基
‑4‑
羟甲基异噁唑溶于无水二氯甲烷,室温搅拌,于0℃氮气氛下滴加三溴化磷(pbr3 2.0eq)。由0℃升至室温搅拌4h后tlc检测反应完全,反应液由无色透明变成浅黄色,加入饱和碳酸氢钠溶液至ph为7
‑
8,二氯甲烷萃取,无水硫酸钠干燥,减压蒸发溶剂,残余物经柱层析分离得到无色透明有刺激性油状物。将4
‑
羟基苯甲酸(1.0eq)和氢氧化钾(koh 2.5eq)溶于乙醇:水为9:1的溶剂中,室温搅拌,向反应体系中滴加化合物上部反应得到的无色透明油状物,加热回流。12h后tlc检测反应完全。冷却至室温后,向反应体系中滴加6m hcl至溶液ph为2
‑
3,并伴有白色固体产生,减压抽滤,烘干,得到的白色固体即为化合物8,收率56%。
[0076]
将化合物8溶于1,2
‑
二氯乙烷,室温搅拌,向搅拌的混合液中依次加入1.2eqhobt、1.5eqedci、0.12eqdmap和3.0eqet3n,室温搅拌一个小时,加入化合物4,加热回流,24h后tlc检测反应完全,反应液由淡黄色变成棕红色,待冷却至室温后加蒸馏水洗涤,二氯甲烷萃取三次,饱和食盐水干燥,无水硫酸钠干燥,减压蒸发溶剂,所得残余物经柱层析色谱分离提纯,后经石油醚:乙酸乙酯体积比为3:1的混合溶剂重结晶得到白色固体z15,收率66%。
[0077]1h nmr(600mhz,dmso
‑
d6)δ12.71(s,1h),8.19(d,j=8.7hz,2h),7.59(d,j=3.0hz,1h),7.56(s,1h),7.54(d,j=4.9hz,1h),7.20(d,j=8.7hz,2h),7.17
–
7.13(m,1h),5.07(s,2h),2.47(s,3h),2.27(s,3h).
[0078]
《实施例2》4
‑
((3,5
‑
二甲基异恶唑
‑4‑
基)甲氧基)
‑
n
‑
(4
‑
(3
‑
氟苯基)噻唑
‑2‑
基)苯甲酰胺的合成(za2)
[0079]
制备方法同化合物za1,以4b和8为原料,制得白色粉末状固体za2,收率71%。
[0080]1h nmr(600mhz,dmso
‑
d6)δ12.68(s,1h),8.19(d,j=8.7hz,2h),7.85(d,j=10.2hz,2h),7.79(d,j=10.5hz,1h),7.53(d,j=7.1hz,1h),7.21(d,j=8.8hz,3h),5.07(s,2h),2.47(s,3h),2.27(s,3h).
[0081]
《实施例3》4
‑
((3,5
‑
二甲基异恶唑
‑4‑
基)甲氧基)
‑
n
‑
(4
‑
(3
‑
甲氧基苯基)噻唑
‑2‑
基)苯甲酰胺的合成(za3)
[0082]
制备方法同化合物za1,以4c和8为原料,制得白色粉末状固体za3,收率65%。
[0083]1h nmr(400mhz,cdcl3)δ10.27(s,1h),7.89(d,j=8.8hz,2h),7.37
‑
7.34(m,2h),7.29(d,j=8.0hz,1h),7.18(s,1h),6.96(d,j=8.8hz,2h),6.84(dd,j=8.1,1.9hz,1h),4.84(s,2h),3.84(s,3h),2.44(s,3h),2.31(s,3h).
[0084]
《实施例4》4
‑
((3,5
‑
二甲基异恶唑
‑4‑
基)甲氧基)
‑
n
‑
(4
‑
(4
‑
氟苯基)噻唑
‑2‑
基)苯甲酰胺的合成(za4)
[0085]
制备方法同化合物za1,以4d和8为原料,制得白色粉末状固体za4,收率62%。
[0086]1h nmr(400mhz,cdcl3)δ10.16(s,1h),7.91(d,j=8.8hz,2h),7.79
–
7.71(m,2h),7.12(s,1h),7.06(t,j=8.7hz,2h),6.99(d,j=8.8hz,2h),4.85(s,2h),2.44(s,3h),2.31(s,3h).
[0087]
《实施例5》4
‑
((3,5
‑
二甲基异恶唑
‑4‑
基)甲氧基)
‑
n
‑
(4
‑
(2
‑
氟苯基)噻唑
‑2‑
基)苯甲酰胺的合成(za5)
[0088]
制备方法同化合物za1,以4e和8为原料,制得白色粉末状固体za5,收率64%。
[0089]1h nmr(400mhz,cdcl3)δ10.76(s,1h),7.91(t,j=7.7hz,1h),7.84
–
7.77(m,2h),
7.45(d,j=2.1hz,1h),7.25
–
7.18(m,1h),7.14
–
7.02(m,2h),6.93
–
6.86(m,2h),4.82(s,2h),2.44(s,3h),2.32(s,3h).
[0090]
《实施例6》n
‑
([4,4'
‑
联噻唑]
‑2‑
基)
‑4‑
((3,5
‑
二甲基异恶唑
‑4‑
基)甲氧基)苯甲酰胺的合成(za6)
[0091]
制备方法同化合物za1,以4f和8为原料,制得白色粉末状固体za6,收率58%。
[0092]1h nmr(600mhz,dmso
‑
d6)δ12.74(s,1h),9.21(s,1h),8.19(d,j=8.5hz,2h),7.91(d,j=1.8hz,1h),7.65(s,1h),7.20(d,j=8.6hz,2h),5.12
–
5.03(m,2h),2.46(s,3h),2.26(s,3h).
[0093]
《实施例7》4
‑
((3,5
‑
二甲基异恶唑
‑4‑
基)甲氧基)
‑
n
‑
(4
‑
(4
‑
(三氟甲氧基)苯基)噻唑
‑2‑
基)苯甲酰胺的合成(za7)
[0094]
制备方法同化合物za1,以4g和8为原料,制得白色粉末状固体za7,收率64%。
[0095]1h nmr(600mhz,dmso
‑
d6)δ12.69(s,1h),8.16(d,j=8.8hz,2h),8.08(d,j=8.7hz,2h),7.77(s,1h),7.46(d,j=8.3hz,2h),7.18(d,j=8.8hz,2h),5.05(s,2h),2.44(s,3h),2.24(s,3h).
[0096]
《实施例8》4
‑
((3,5
‑
二甲基异恶唑
‑4‑
基)甲氧基)
‑
n
‑
(4
‑
(5
‑
甲基噻吩
‑2‑
基)噻唑
‑2‑
基)苯甲酰胺的合成(za8)
[0097]
制备方法同化合物za1,以4h和8为原料,制得白色粉末状固体za8,收率45%。
[0098]1h nmr(400mhz,dmso
‑
d6)δ12.64(s,1h),8.14(d,j=8.9hz,2h),7.40(s,1h),7.32(d,j=3.5hz,1h),7.16(d,j=8.9hz,2h),6.80(dd,j=3.5,1.1hz,1h),5.04(s,2h),2.46(d,j=0.4hz,3h),2.43(s,3h),2.23(s,3h).
[0099]
《实施例9》n
‑
(4
‑
(5
‑
溴噻吩
‑2‑
基)噻唑
‑2‑
基)
‑4‑
((3,5
‑
二甲基异恶唑
‑4‑
基)甲氧基)苯甲酰胺的合成(za9)
[0100]
制备方法同化合物za1,以4i和8为原料,制得白色粉末状固体za9,收率58%。
[0101]1h nmr(600mhz,dmso
‑
d6)δ12.81(s,1h),8.25(d,j=8.8hz,2h),7.71(s,1h),7.51(d,j=3.9hz,1h),7.35(d,j=3.9hz,1h),7.27(d,j=8.8hz,2h),5.14(s,2h),2.54(s,3h),2.34(s,3h).
[0102]
《实施例10》n
‑
(4
‑
(5
‑
氯噻吩
‑2‑
基)噻唑
‑2‑
基)
‑4‑
((3,5
‑
二甲基异恶唑
‑4‑
基)甲氧基)苯甲酰胺的合成(za10)
[0103]
制备方法同化合物za1,以4j和8为原料,制得白色粉末状固体za10,收率53%。
[0104]1h nmr(600mhz,dmso
‑
d6)δ12.74(s,1h),8.18(d,j=8.8hz,2h),7.63(s,1h),7.47(d,j=3.9hz,1h),7.19(d,j=8.8hz,2h),7.18(d,j=3.9hz,1h),5.07(s,2h),2.47(s,3h),2.27(s,3h).
[0105]
《实施例11》4
‑
((3,5
‑
二甲基异恶唑
‑4‑
基)甲氧基)
‑
n
‑
(4
‑
(呋喃
‑2‑
基)噻唑
‑2‑
基)苯甲酰胺的合成(za11)
[0106]
制备方法同化合物za1,以4k和8为原料,制得白色粉末状固体za11,收率68%。
[0107]1h nmr(600mhz,dmso
‑
d6)δ12.75(s,1h),8.18(d,j=8.9hz,2h),7.78(d,j=1.0hz,1h),7.43(s,1h),7.20(d,j=8.9hz,2h),6.77(d,j=3.2hz,1h),6.64(dd,j=3.3,1.8hz,1h),5.07(s,2h),2.47(s,3h),2.27(s,3h).
[0108]
《实施例12》4
‑
((3,5
‑
二甲基异恶唑
‑4‑
基)甲氧基)
‑
n
‑
(4
‑
(4
‑
(三氟甲基)苯基)噻
唑
‑2‑
基)苯甲酰胺的合成(za12)
[0109]
制备方法同化合物za1,以4l和8为原料,制得白色粉末状固体za12,收率49%。
[0110]1h nmr(400mhz,dmso
‑
d6)δ12.76(s,1h),8.21(t,j=7.4hz,4h),7.96(s,1h),7.86(d,j=8.1hz,2h),7.21(d,j=8.6hz,2h),5.08(s,2h),2.47(s,3h),2.27(s,3h).
[0111]
《实施例13》n
‑
(4
‑
(3
‑
氰基苯基)噻唑
‑2‑
基)
‑4‑
((3,5
‑
二甲基异恶唑
‑4‑
基)甲氧基)苯甲酰胺的合成(za13)
[0112]
制备方法同化合物za1,以4m和8为原料,制得白色粉末状固体za13,收率62%。
[0113]1h nmr(400mhz,dmso
‑
d6)δ12.71(s,1h),8.43(s,1h),8.32(d,j=8.0hz,1h),8.19(d,j=8.8hz,2h),7.96(s,1h),7.84(d,j=7.7hz,1h),7.71(t,j=7.8hz,1h),7.21(d,j=8.8hz,2h),5.08(s,2h),2.47(s,3h),2.27(s,3h).
[0114]
《实施例14》n
‑
(4
‑
(3
‑
氰基苯基)噻唑
‑2‑
基)
‑4‑
((3,5
‑
二甲基异恶唑
‑4‑
基)甲氧基)苯甲酰胺的合成(za14)
[0115]
制备方法同化合物za1,以4n和8为原料,制得白色粉末状固体za14,收率57%。
[0116]1h nmr(600mhz,dmso
‑
d6)δ12.79(s,1h),8.20(d,j=8.8hz,2h),8.00(d,j=7.9hz,1h),7.90(d,j=5.8hz,2h),7.79(s,1h),7.48
–
7.36(m,2h),7.21(d,j=8.9hz,2h),5.08(s,2h),2.47(s,3h),2.27(s,3h).
[0117]
《实施例15》4
‑
((3,5
‑
二甲基异恶唑
‑4‑
基)甲氧基)
‑
n
‑
(4
‑
(3
‑
(三氟甲基)苯基)噻唑
‑2‑
基)苯甲酰胺的合成(za15)
[0118]
制备方法同化合物za1,以4o和8为原料,制得白色粉末状固体za15,收率60%。
[0119]1h nmr(400mhz,dmso
‑
d6)δ12.68(s,1h),8.36(s,1h),8.28(t,j=3.7hz,1h),8.20(d,j=8.8hz,2h),7.94(s,1h),7.70(t,j=6.4hz,2h),7.20(d,j=8.9hz,2h),5.06(s,2h),2.46(s,3h),2.26(s,3h).
[0120]
《实施例16》n
‑
(4
‑
(苯并呋喃
‑2‑
基)噻唑
‑2‑
基)
‑4‑
((3,5
‑
二甲基异恶唑
‑4‑
基)甲氧基)苯甲酰胺的合成(za16)
[0121]
制备方法同化合物za1,以4p和8为原料,制得白色粉末状固体za16,收率66%。
[0122]1h nmr(400mhz,dmso
‑
d6)δ12.88(s,1h),8.23(d,j=8.8hz,2h),7.78(s,1h),7.76(d,j=7.8hz,1h),7.69(d,j=8.1hz,1h),7.40(dd,j=11.2,4.2hz,1h),7.34(t,j=7.4hz,1h),7.24(d,j=9.1hz,3h),5.10(s,2h),2.49(s,3h),2.29(s,3h).
[0123]
《实施例17》4
‑
((3,5
‑
二甲基异恶唑
‑4‑
基)甲氧基)
‑
n
‑
(4
‑
(噻吩
‑2‑
基)
‑
1h
‑
咪唑
‑2‑
基)苯甲酰胺的合成(zb1)
[0124]
将2
‑
乙酰基噻吩和tsoh(0.1eq)溶于二氯甲烷中,室温搅拌,分批次加入nbs(1.0eq),加热回流,12h后tlc检测反应完全,反应液由浅黄色变为棕红色。冷却至室温后加入适量饱和食盐水洗涤,二氯甲烷萃取,无水硫酸钠干燥,减压蒸发溶剂,残余物经柱层析分离得到棕红色油状物2。将油状物2溶于dmf,向搅拌的溶液中加入3a(1
‑
乙酰基胍,3.0eq),升温至60℃搅拌。12h后tlc检测反应完全,反应液由浅黄色变成棕红色。冷却后用大量饱和食盐水洗去dmf,乙酸乙酯萃取,无水硫酸钠干燥,减压蒸发溶剂,残余物经柱层析分离得到淡黄色固体。将所分离的淡黄色固体溶解在甲醇中,向搅拌的混合液中加入8m hcl,加热回流。4h后tlc检测反应完全,冷却至室温,6m naoh调节溶液ph值为10,乙酸乙酯萃取,无水硫酸钠干燥,减压蒸发溶剂,残余物经柱层析分离得到淡黄色固体4'a,收率
37%。
[0125]
制备方法同化合物za1,以4'a和8为原料,制得白色粉末状固体zb1,收率47%。
[0126]1h nmr(600mhz,dmso
‑
d6)δ11.98(s,1h),11.60(s,1h),8.13(d,j=8.5hz,2h),7.36(d,j=4.1hz,1h),7.32(s,1h),7.26(s,1h),7.17(d,j=8.5hz,2h),7.07(s,1h),5.06(s,2h),2.47(s,3h),2.27(s,3h).
[0127]
《实施例18》n
‑
(苯并[d]恶唑
‑2‑
基)
‑4‑
((3,5
‑
二甲基异恶唑
‑4‑
基)甲氧基)苯甲酰胺的合成(ze1)
[0128]
制备方法同化合物za1,以9a和8为原料,制得白色粉末状固体ze1,收率37%。
[0129]1h nmr(600mhz,dmso
‑
d6)δ12.06(s,1h),8.10(d,j=8.4hz,2h),7.67(d,j=7.7hz,1h),7.63(d,j=7.1hz,1h),7.36(dd,j=13.6,7.6hz,2h),7.19(d,j=8.6hz,2h),5.07(s,2h),2.47(s,3h),2.27(s,3h).
[0130]
《实施例19》4
‑
((3,5
‑
二甲基异恶唑
‑4‑
基)甲氧基)
‑
n
‑
(5
‑
氟苯并[d]噻唑
‑2‑
基)苯甲酰胺的合成(ze2)
[0131]
制备方法同化合物za1,以9b和8为原料,制得白色粉末状固体ze2,收率55%。
[0132]1h nmr(600mhz,dmso
‑
d6)δ12.86(s,1h),8.17(d,j=8.8hz,2h),8.05(dd,j=8.5,5.5hz,1h),7.61(d,j=9.6hz,1h),7.23(td,j=9.2,2.4hz,1h),7.19(d,j=8.8hz,2h),5.05(s,2h),2.44(s,3h),2.23(s,3h).
[0133]
《实施例20》4
‑
((3,5
‑
二甲基异恶唑
‑4‑
基)甲氧基)
‑
n
‑
(5
‑
甲氧基苯并[d]噻唑
‑2‑
基)苯甲酰胺的合成(ze3)
[0134]
制备方法同化合物za1,以9c和8为原料,制得白色粉末状固体ze3,收率58%。
[0135]1h nmr(600mhz,dmso
‑
d6)δ12.75(s,1h),8.19(d,j=8.7hz,2h),7.90(d,j=8.7hz,1h),7.32(s,1h),7.21(d,j=8.8hz,2h),7.01(dd,j=8.7,2.3hz,1h),5.08(s,2h),3.87(s,3h),2.47(s,3h),2.27(s,3h).
[0136]
《实施例21》4
‑
((3,5
‑
二甲基异恶唑
‑4‑
基)甲氧基)
‑
n
‑
(6
‑
(三氟甲氧基)苯并[d]噻唑
‑2‑
基)苯甲酰胺的合成(ze4)
[0137]
制备方法同化合物za1,以9d和8为原料,制得白色粉末状固体ze4,收率43%。
[0138]1h nmr(600mhz,dmso
‑
d6)δ12.88(s,1h),8.24
–
8.13(m,3h),7.87(d,j=8.7hz,1h),7.46(d,j=8.8hz,1h),7.20(d,j=8.8hz,2h),5.06(s,2h),2.44(s,3h),2.24(s,3h).《实施例22》4
‑
((3,5
‑
二甲基异恶唑
‑4‑
基)甲氧基)
‑
n
‑
(6
‑
氟苯并[d]噻唑
‑2‑
基)苯甲酰胺的合成(ze5)
[0139]
制备方法同化合物za1,以9e和8为原料,制得白色粉末状固体ze5,收率38%。
[0140]1h nmr(600mhz,dmso
‑
d6)δ12.78(s,1h),8.17(d,j=8.8hz,2h),7.93(dd,j=8.6,2.5hz,1h),7.79(dd,j=8.7,4.7hz,1h),7.32(td,j=9.1,2.6hz,1h),7.19(d,j=8.8hz,2h),5.05(s,2h),2.44(s,3h),2.24(s,3h).
[0141]
《实施例23》n
‑
(6
‑
氯苯并[d]噻唑
‑2‑
基)
‑4‑
((3,5
‑
二甲基异恶唑
‑4‑
基)甲氧基)苯甲酰胺的合成(ze6)
[0142]
制备方法同化合物za1,以9f和8为原料,制得白色粉末状固体ze6,收率63%。
[0143]1h nmr(600mhz,dmso
‑
d6)δ12.84(s,1h),8.17(d,j=8.5hz,3h),7.77(d,j=8.6hz,1h),7.49(dd,j=8.6,2.1hz,1h),7.19(d,j=8.8hz,2h),5.05(s,2h),2.44(s,3h),
2.24(s,3h).
[0144]
《实施例24》n
‑
(5,6
‑
二甲基苯并[d]噻唑
‑2‑
基)
‑4‑
((3,5
‑
二甲基异恶唑
‑4‑
基)甲氧基)苯甲酰胺的合成(ze7)
[0145]
制备方法同化合物za1,以9g和8为原料,制得白色粉末状固体ze7,收率54%。
[0146]1h nmr(600mhz,dmso
‑
d6)δ12.63(s,1h),8.15(d,j=8.8hz,2h),7.74(s,1h),7.57(s,1h),7.17(d,j=8.8hz,2h),5.04(s,2h),2.43(s,3h),2.34(d,j=6.8hz,6h),2.23(s,3h).
[0147]
《实施例25》4
‑
((3,5
‑
二甲基异恶唑
‑4‑
基)甲氧基)
‑3‑
甲氧基
‑
n
‑
(4
‑
(噻吩
‑2‑
基)噻唑
‑2‑
基)苯甲酰胺的合成(zc1)
[0148]
制备方法同化合物za1,以8a和4为原料,制得白色粉末状固体zc1,收率36%。
[0149]1h nmr(600mhz,dmso
‑
d6)δ12.74(s,1h),7.83(d,j=3.9hz,2h),7.59(d,j=3.5hz,1h),7.56
‑
7.3(m,2h),7.28(d,j=7.8hz,1h),7.18
–
7.12(m,1h),5.04(d,j=1.3hz,2h),3.90(s,3h),2.45(s,3h),2.26(s,3h).
[0150]
《实施例26》4
‑
((3,5
‑
二甲基异恶唑
‑4‑
基)甲氧基)
‑3‑
甲基
‑
n
‑
(4
‑
(噻吩
‑2‑
基)噻唑
‑2‑
基)苯甲酰胺的合成(zc2)
[0151]
制备方法同化合物zc2,以8b和4为原料,制得白色粉末状固体zc2,收率62%。
[0152]1h nmr(400mhz,dmso
‑
d6)δ12.59(s,1h),8.13
–
7.95(m,2h),7.54(s,1h),7.49(s,2h),7.29
–
7.00(m,2h),5.03(s,2h),2.43(s,3h),2.25(s,3h),2.19(s,3h).
[0153]
《实施例27》4
‑
((3,5
‑
二甲基异恶唑
‑4‑
基)甲氧基)
‑3‑
氟
‑
n
‑
(4
‑
(噻吩
‑2‑
基)噻唑
‑2‑
基)苯甲酰胺的合成(zc3)
[0154]
制备方法同化合物za1,以8c和4为原料,制得白色粉末状固体zc3,收率63%。
[0155]1h nmr(600mhz,dmso
‑
d6)δ12.80(s,1h),8.11
–
8.04(m,2h),7.62
–
7.56(m,2h),7.55(d,j=4.9hz,1h),7.51(t,j=8.4hz,1h),7.17
‑
7.15(m,1h),5.17(s,2h),2.47(s,3h),2.27(s,3h).
[0156]
《实施例28》4
‑
((3,5
‑
二甲基异恶唑
‑4‑
基)甲氧基)
‑
n
‑
(4
‑
(噻吩
‑2‑
基)噻唑
‑2‑
基)
‑3‑
(三氟甲基)苯甲酰胺的合成(zc4)
[0157]
制备方法同化合物za1,以8d和4为原料,制得白色粉末状固体zc4,收率65%。
[0158]1h nmr(400mhz,dmso
‑
d6)δ12.97(s,1h),8.50(d,j=11.4hz,2h),7.63
‑
7.59(m,3h),7.55(d,j=5.0hz,1h),7.17
‑
7.15(m,1h),5.24(s,2h),2.49(s,3h),2.26(s,3h).
[0159]
《实施例29》6
‑
((3,5
‑
二甲基异恶唑
‑4‑
基)甲氧基)
‑
n
‑
(4
‑
(噻吩
‑2‑
基)噻唑
‑2‑
基)烟酰胺的合成(zc5)
[0160]
制备方法同化合物zc5,以8e和4为原料,制得白色粉末状固体zc5,收率57%。
[0161]1h nmr(600mhz,dmso
‑
d6)δ12.64(s,1h),8.79(d,j=2.5hz,1h),8.14(dd,j=9.6,2.6hz,1h),7.58(d,j=3.5hz,1h),7.55(d,j=1.2hz,1h),7.54(d,j=5.0hz,1h),7.17
–
7.12(m,1h),6.53(d,j=9.5hz,1h),5.00(s,2h),2.47(s,3h),2.23(s,3h).
[0162]
《实施例30》3
‑
氯
‑4‑
((3,5
‑
二甲基异恶唑
‑4‑
基)甲氧基)
‑
n
‑
(4
‑
(噻吩
‑2‑
基)噻唑
‑2‑
基)苯甲酰胺的合成(zc6)
[0163]
制备方法同化合物za1,以8f和4为原料,制得白色粉末状固体zc6,收率59%。
[0164]1h nmr(600mhz,dmso
‑
d6)δ12.78(s,1h),8.28(d,j=2.2hz,1h),8.17(dd,j=8.7,
2.2hz,1h),7.56(d,j=3.5hz,1h),7.55(s,1h),7.52(d,j=5.0hz,1h),7.46(d,j=8.8hz,1h),7.13(dd,j=5.0,3.7hz,1h),5.16(s,2h),2.46(s,3h),2.27(s,3h).
[0165]
《实施例31》4'
‑
((3,5
‑
二甲基异恶唑
‑4‑
基)甲氧基)
‑
n
‑
(4
‑
(噻吩
‑2‑
基)噻唑
‑2‑
基)
‑
[1,1'
‑
联苯]
‑4‑
甲酰胺的合成(zc7)
[0166]
制备方法同化合物za1,以8g和4为原料,制得白色粉末状固体zc7,收率68%。
[0167]1h nmr(600mhz,dmso
‑
d6)δ12.85(s,1h),8.21(d,j=8.1hz,2h),7.83(d,j=8.1hz,2h),7.76(d,j=8.4hz,2h),7.57(d,j=2.7hz,1h),7.56(s,1h),7.52(d,j=4.9hz,1h),7.14(t,j=8.4hz,3h),4.99(s,2h),2.44(s,3h),2.24(s,3h).
[0168]
《实施例32》6
‑
((3,5
‑
二甲基异恶唑
‑4‑
基)甲氧基)
‑
n
‑
(4
‑
(噻吩
‑2‑
基)噻唑
‑2‑
基)
‑2‑
萘甲酰胺的合成(zc8)
[0169]
制备方法同化合物z15,以8h和4为原料,制得白色粉末状固体zc8,收率56%。
[0170]1h nmr(600mhz,dmso
‑
d6)δ12.87(s,1h),8.78(s,1h),8.13(d,j=8.6hz,1h),8.01(d,j=9.0hz,1h),7.95(d,j=8.6hz,1h),7.57(dd,j=7.0,4.0hz,3h),7.52(d,j=5.0hz,1h),7.33(dd,j=8.9,2.2hz,1h),7.16
–
7.11(m,1h),5.11(s,2h),2.47(s,3h),2.26(s,3h).
[0171]
《实施例33》4
‑
((3,5
‑
二甲基异恶唑
‑4‑
基)甲氧基)
‑3‑
甲酰基
‑
n
‑
(4
‑
(噻吩
‑2‑
基)噻唑
‑2‑
基)苯甲酰胺的合成(zc9)
[0172]
制备方法同化合物za1,以8i和4为原料,制得白色粉末状固体zc9,收率66%。
[0173]1h nmr(600mhz,dmso
‑
d6)δ12.95(s,1h),10.34(s,1h),8.56(d,j=2.3hz,1h),8.49(dd,j=8.8,2.3hz,1h),7.58(dd,j=9.1,5.4hz,3h),7.55(d,j=4.9hz,1h),7.20
–
7.13(m,1h),5.26(s,2h),2.50(s,3h),2.30(s,3h).
[0174]
《实施例34》3
‑
氰基
‑4‑
((3,5
‑
二甲基异恶唑
‑4‑
基)甲氧基)
‑
n
‑
(4
‑
(噻吩
‑2‑
基)噻唑
‑2‑
基)苯甲酰胺的合成(zc10)
[0175]
制备方法同化合物z15,以8j和4为原料,制得白色粉末状固体zc10,收率53%。
[0176]1h nmr(600mhz,dmso
‑
d6)δ12.84(s,1h),8.56(d,j=2.2hz,1h),8.44(dd,j=8.9,2.2hz,1h),7.57(t,j=4.5hz,3h),7.52(d,j=4.7hz,1h),7.16
–
7.10(m,1h),5.25(s,2h),2.47(s,3h),2.28(s,3h).
[0177]
《实施例35》4
‑
((3,5
‑
二甲基异恶唑
‑4‑
基)甲氧基)
‑3‑
硝基
‑
n
‑
(4
‑
(噻吩
‑2‑
基)噻唑
‑2‑
基)苯甲酰胺的合成(zc11)
[0178]
制备方法同化合物za1,以8k和4为原料,制得白色粉末状固体zc11,收率57%。
[0179]1h nmr(400mhz,dmso
‑
d6)δ12.96(s,1h),8.71(d,j=2.3hz,1h),8.46(dd,j=8.9,2.3hz,1h),7.68(d,j=9.0hz,1h),7.59
–
7.54(m,2h),7.52(dd,j=5.0,1.1hz,1h),7.13(dd,j=5.0,3.6hz,1h),5.26(s,2h),2.45(s,3h),2.25(s,3h).
[0180]
《实施例36》4
‑
((3,5
‑
二甲基异恶唑
‑4‑
基)甲氧基)
‑3‑
(羟甲基)
‑
n
‑
(4
‑
(噻吩
‑2‑
基)t
‑
噻唑
‑2‑
基)苯甲酰胺的合成(zc12)
[0181]
zc12由zc9经硼氢化钠还原得到。
[0182]1h nmr(600mhz,dmso
‑
d6)δ12.67(s,1h),8.19(d,j=1.8hz,1h),8.15(dd,j=8.6,2.1hz,1h),7.55(d,j=3.4hz,1h),7.51(d,j=4.0hz,2h),7.24(d,j=8.6hz,1h),7.15
–
7.09(m,1h),5.19(t,j=5.4hz,1h),5.06(s,2h),4.51(d,j=5.4hz,2h),2.44(s,3h),2.25
(s,3h).
[0183]
《实施例37》4
‑
((1h
‑
吡唑
‑1‑
基)甲氧基)
‑
n
‑
(4
‑
(噻吩
‑2‑
基)噻唑
‑2‑
基)苯甲酰胺的合成(zd1)
[0184]
将商业可得的化合物1
‑
吡唑甲醇(5a)溶于氯化亚砜,加热回流。4h后tlc检测反应完成,减压蒸发溶剂,得到残余物6a。将化合物14和碳酸铯(cs2co
3 2.5eq)溶于dmf中,室温搅拌,向反应体系中加入化合物6a,70℃加热搅拌。12h后tlc检测反应完全。冷却至室温后,用大量饱和食盐水将dmf洗去,乙酸乙酯萃取,减压蒸发溶剂,残余物柱层析分离纯化得白色固体。将分离得到的固体和氢氧化钠(4eq)溶解在甲醇:水=9:1的溶剂中,加热回流,2h后tlc检测反应完全。反应液冷却至室温,滴加6m盐酸,调至溶液ph在4
‑
6,有白色固体析出。减压抽滤烘干,得到白色固体化合物15a。
[0185]
制备方法同化合物z15,以15a和4为原料,制得白色粉末状固体zd1,收率46%。
[0186]1h nmr(600mhz,dmso
‑
d6)δ12.73(s,1h),8.19
–
8.14(m,2h),8.09(d,j=2.3hz,1h),7.64(d,j=1.5hz,1h),7.59(dd,j=3.5,1.0hz,1h),7.57(s,1h),7.55(dd,j=5.0,0.9hz,1h),7.31(d,j=8.9hz,2h),7.16(dd,j=5.0,3.6hz,1h),6.42
–
6.38(m,1h),6.24(s,2h)
[0187]
《实施例38》4
‑
((1h
‑
吲哚
‑1‑
基)甲基)
‑
n
‑
(4
‑
(噻吩
‑2‑
基)噻唑
‑2‑
基)苯甲酰胺的合成(zd2)
[0188]
将吲哚和碳酸铯(cs2co
3 2.5eq)溶于dmf中,室温搅拌,向反应体系中加入化合物14a,70℃加热搅拌。12h后tlc检测反应完全。冷却至室温后,用大量饱和食盐水将dmf洗去,乙酸乙酯萃取,减压蒸发溶剂,残余物柱层析分离纯化得白色固体。将分离得到的固体和氢氧化钠(4eq)溶解在甲醇:水=9:1的溶剂中,加热回流,2h后tlc检测反应完全。反应液冷却至室温,滴加6m盐酸,调至溶液ph在4
‑
6,有白色固体析出。减压抽滤烘干,得到白色固体化合物15c。
[0189]
制备方法同化合物za1,以15c和4为原料,制得白色粉末状固体zd2,收率76%。
[0190]1h nmr(600mhz,dmso
‑
d6)δ12.82(s,1h),8.09(d,j=8.2hz,2h),7.62(d,j=7.8hz,1h),7.59(dd,j=5.6,3.4hz,2h),7.57(s,1h),7.54(d,j=5.0hz,1h),7.48(d,j=8.2hz,1h),7.34(d,j=8.2hz,2h),7.15(d,j=3.7hz,1h),7.14(t,j=5.3hz,1h),7.07(t,j=7.4hz,1h),6.56(d,j=2.9hz,1h),5.58(s,2h).
[0191]
《实施例39》4
‑
(吲哚
‑1‑
基甲基)
‑
n
‑
(4
‑
(噻吩
‑2‑
基)噻唑
‑2‑
基)苯甲酰胺的合成(zd3)
[0192]
制备方法同化合物zd2,收率64%。
[0193]1h nmr(600mhz,dmso
‑
d6)δ12.84(s,1h),8.16(d,j=8.0hz,2h),7.63
–
7.57(m,2h),7.57
–
7.50(m,3h),7.20
–
7.13(m,1h),7.10(d,j=7.0hz,1h),7.02(t,j=7.6hz,1h),6.63(t,j=7.3hz,1h),6.60(d,j=7.8hz,1h),4.39(s,2h),3.34(t,j=8.3hz,2h),2.96(t,j=8.2hz,2h).
[0194]
《实施例40》4
‑
(2
‑
(1h
‑
吲哚
‑1‑
基)乙氧基)
‑
n
‑
(4
‑
(噻吩
‑2‑
基)噻唑
‑2‑
基)苯甲酰胺的合成(zd4)
[0195]
将吲哚和碳酸铯(cs2co
3 2.5eq)溶于dmf中,室温搅拌,向反应体系中加入化合物2
‑
溴乙醇,70℃加热搅拌。12h后tlc检测反应完全。冷却至室温后,用大量饱和食盐水将dmf
洗去,乙酸乙酯萃取,减压蒸发溶剂,残余物柱层析分离纯化得白色固体。将分离得到的固体和氢氧化钠(4eq)溶解在甲醇:水=9:1的溶剂中,加热回流,2h后tlc检测反应完全。反应液冷却至室温,滴加6m盐酸,调至溶液ph在4
‑
6,有白色固体析出。减压抽滤烘干,得到白色固体化合物16,收率85%。
[0196]
将化合物16溶于甲苯,室温搅拌,向搅拌的混合液中依次加入n
‑
甲基咪唑(nmi1.5eq)、对甲苯磺酰氯(tscl 1.5eq)、三乙胺(1.5eq),室温搅拌,1h后tlc检测反应完全。加入甲醇,减压蒸发溶剂,残余物经柱层析色谱分离得到白色固体17。将化合物16和碳酸铯(cs2co
3 2.5eq)溶于dmf中,室温搅拌,向反应体系中加入尼泊金甲酯(化合物14),70℃加热搅拌。12h后tlc检测反应完全。冷却至室温后,用大量饱和食盐水将dmf洗去,乙酸乙酯萃取,减压蒸发溶剂,残余物经柱层析分离纯化得白色固体。将分离得到的固体和氢氧化钠(4eq)溶解在甲醇:水=9:1的溶剂中,加热回流,2h后tlc检测反应完全。反应液冷却至室温,滴加6m盐酸,调至溶液ph在4
‑
6,有白色固体析出。减压抽滤烘干,得到白色固体化合物15e,收率81%。
[0197]
制备方法同化合物za1,以15e和4为原料,制得白色粉末状固体zd4,收率73%。
[0198]1h nmr(400mhz,dmso
‑
d6)δ12.64(s,1h),8.09(d,j=8.9hz,2h),7.58(d,j=8.3hz,1h),7.55(d,j=3.4hz,1h),7.53(d,j=10.2hz,2h),7.50(s,1h),7.45(d,j=3.1hz,1h),7.16(t,j=7.7hz,1h),7.12(dd,j=5.0,3.7hz,1h),7.03(dd,j=8.0,4.2hz,3h),6.46(d,j=2.9hz,1h),4.61(t,j=5.1hz,2h),4.41(t,j=5.2hz,2h).
[0199]
《实施例41》4
‑
(2
‑
(吲哚
‑1‑
基)乙氧基)
‑
n
‑
(4
‑
(噻吩
‑2‑
基)噻唑
‑2‑
基)苯甲酰胺的合成(zd5)
[0200]
制备方法同化合物zd4,收率63%。
[0201]1h nmr(600mhz,dmso
‑
d6)δ12.66(s,1h),8.13(d,j=8.9hz,2h),7.55(dd,j=3.5,1.0hz,1h),7.53(s,1h),7.51(dd,j=5.0,1.0hz,1h),7.14
–
7.12(m,1h),7.11(d,j=8.8hz,2h),7.04(d,j=7.1hz,1h),7.00(t,j=7.7hz,1h),6.62
–
6.55(m,2h),4.30(t,j=5.6hz,2h),3.51
–
3.43(m,4h),2.90(t,j=8.4hz,2h).
[0202]
《实施例42》4
‑
(((5
‑
甲基异恶唑
‑4‑
基)甲氧基)甲基)
‑
n
‑
(4
‑
(噻吩
‑2‑
基)噻唑
‑2‑
基)苯甲酰胺的合成(zln1)
[0203]
用无水thf将化合物5溶解,室温搅拌,氮气氛下加入氢化钠(nah 1.5eq)搅拌1h,后将四丁基碘化铵(tbai 0.12eq)和化合物6依次加入搅拌的混合物中。12h后tlc检测反应完全。饱和氯化铵水溶液淬灭剩余的氢化钠,乙酸乙酯萃取,无水硫酸钠干燥,减压蒸发溶剂,残余物经柱层析色谱分离得到淡黄色固体。将分离得到的固体和氢氧化钠(4eq)溶解在甲醇:水=9:1的溶剂中,加热回流,2h后tlc检测反应完全。反应液冷却至室温,滴加6m盐酸,调至溶液ph在4
‑
6,有白色固体析出。减压抽滤烘干,得到白色固体化合物8'e,收率37%。
[0204]
制备方法同化合物za1,以8'e和4为原料,制得白色粉末状固体zln1,收率66%。
[0205]1h nmr(600mhz,dmso
‑
d6)δ12.83(s,1h),8.12(d,j=8.1hz,2h),7.56(d,j=3.4hz,1h),7.55(s,1h),7.52(d,j=5.0hz,1h),7.49(d,j=8.1hz,2h),7.15
–
7.11(m,1h),4.57(s,2h),4.38(s,2h),2.37(s,3h),2.21(s,3h).
[0206]
《实施例43》4
‑
(2
‑
((3,5
‑
二甲基异恶唑
‑4‑
基)甲氧基)乙氧基)
‑
n
‑
(4
‑
(噻吩
‑2‑
基)噻唑
‑2‑
基)苯甲酰胺的合成(zln2)
[0207]
将化合物14和碳酸钾(k2co
3 2.5eq)溶于dmf中,室温搅拌,向反应体系中加入化合物17a,70℃加热搅拌。12h后tlc检测反应完全。冷却至室温后,用大量饱和食盐水将dmf洗去,乙酸乙酯萃取,减压蒸发溶剂,残余物柱层析分离纯化得白色固体18a,收率85%。
[0208]
制备方法同8'e,以18a和6为原料,制得白色粉末状固体8'a,收率49%。
[0209]
制备方法同化合物z15,以8'a和4为原料,制得白色粉末状固体zln3,收率54%。
[0210]1h nmr(600mhz,dmso
‑
d6)δ12.66(s,1h),8.13(d,j=8.7hz,2h),7.56(d,j=3.2hz,1h),7.53(s,1h),7.51(d,j=4.9hz,1h),7.16
–
7.10(m,1h),7.08(d,j=8.8hz,2h),4.38(s,2h),4.25
–
4.18(m,2h),3.77
–
3.68(m,2h),2.37(s,3h),2.19(s,3h).
[0211]
《实施例44》4
‑
(3
‑
((3,5
‑
二甲基异恶唑
‑4‑
基)甲氧基)丙氧基)
‑
n
‑
(4
‑
(噻吩
‑2‑
基)噻唑
‑2‑
基)苯甲酰胺的合成(zln3)
[0212]
制备方法同化合物zln2,以8'b和4为原料,制得白色粉末状固体zln3,收率67%。
[0213]1h nmr(600mhz,dmso
‑
d6)δ12.69(s,1h),8.15(d,j=8.7hz,2h),7.59(d,j=2.7hz,1h),7.57
–
7.49(m,2h),7.15(dd,j=4.6,3.8hz,1h),7.08(d,j=8.7hz,2h),4.32(s,2h),4.15(t,j=6.1hz,2h),3.56(t,j=6.1hz,2h),2.37(s,3h),2.18(s,3h),2.08
–
1.95(m,2h).
[0214]
《实施例45》4
‑
(4
‑
((3,5
‑
二甲基异恶唑
‑4‑
基)甲氧基)丁氧基)
‑
n
‑
(4
‑
(噻吩
‑2‑
基)噻唑
‑2‑
基)苯甲酰胺的合成(zln4)
[0215]
制备方法同化合物zln2,以8'c和4为原料,制得白色粉末状固体zln4,收率46%。
[0216]1h nmr(600mhz,dmso
‑
d6)δ12.69(s,1h),8.15(d,j=8.7hz,2h),7.59(d,j=3.2hz,1h),7.56(s,1h),7.54(d,j=5.0hz,1h),7.08(d,j=8.8hz,2h),4.30(s,2h),4.11(t,j=6.4hz,2h),3.46(t,j=6.3hz,2h),2.38(s,3h),2.21(d,j=5.6hz,3h),1.88
–
1.77(m,2h),1.70(dt,j=12.9,6.3hz,3h).
[0217]
《实施例46》4
‑
(((5
‑
((3,5
‑
二甲基异恶唑
‑4‑
基)甲氧基)戊基)氧基)
‑
n
‑
(4
‑
(噻吩
‑2‑
基)噻唑
‑
醇
‑2‑
基)苯甲酰胺的合成(zln5)
[0218]
制备方法同化合物zln2,以8'd和4为原料,制得白色粉末状固体zln5,收率42%。
[0219]1h nmr(600mhz,dmso
‑
d6)δ12.64(s,1h),8.12(d,j=8.8hz,2h),7.55(d,j=3.4hz,1h),7.52(s,1h),7.51(d,j=5.0hz,1h),7.12(dd,j=4.9,3.7hz,1h),7.05(d,j=8.8hz,2h),4.26(s,2h),4.06(t,j=6.4hz,2h),3.38(t,j=6.3hz,3h),2.35(s,3h),2.17(s,3h),1.81
–
1.69(m,2h),1.65
–
1.53(m,2h),1.51
–
1.38(m,2h).
[0220]
《实施例47》4
‑
((3,5
‑
二甲基异恶唑
‑4‑
基)甲氧基)
‑
n
‑
(4
‑
(4
‑
甲氧基苯基)噻唑
‑2‑
基)
‑3‑
甲基苯甲酰胺的合成(zac1)
[0221]
将商业可得的对甲氧基苯乙酮和对甲苯磺酸(tsoh,0.1eq)溶于无水二氯甲烷中,室温搅拌,分批次加入n
‑
溴代丁二酰亚胺(nbs,1.0eq),加热回流,12h后tlc检测反应完全,反应液由浅黄色变为棕红色。冷却至室温后加入适量饱和食盐水洗涤,二氯甲烷萃取,无水硫酸钠干燥,减压蒸发溶剂,残余物经柱层析分离得到棕红色油状物。将硫脲(1.2eq)溶于乙醇,室温搅拌,加入棕红色油状物的乙醇溶液,加热回流。2h后tlc检测反应完全,反应液由浅黄色变成深黄色。冷却后减压蒸发溶剂,加入适量饱和碳酸氢钠溶液洗涤,乙酸乙酯萃取,无水硫酸钠干燥,减压蒸发溶剂,残余物经柱层析分离得到淡黄色固体4
‑
(4
‑
甲氧基苯
基)噻唑
‑2‑
胺,收率60%。
[0222]
制备方法同化合物za1,以4
‑
(4
‑
甲氧基苯基)噻唑
‑2‑
胺和8b为原料,制得白色粉末状固体zac1,收率,68%。
[0223]1h nmr(600mhz,dmso
‑
d6)δ12.51(s,1h),8.07(d,j=8.4hz,1h),8.02(s,1h),7.89(d,j=8.3hz,2h),7.49(s,1h),7.24(d,j=8.5hz,1h),7.01(d,j=8.3hz,2h),5.05(s,2h),3.80(s,3h),2.45(s,3h),2.26(s,3h),2.20(s,3h).
[0224]
《实施例48》4
‑
((3,5
‑
二甲基异恶唑
‑4‑
基)甲氧基)
‑
n
‑
(5
‑
甲氧基苯并[d]噻唑
‑2‑
基)
‑3‑
甲基苯甲酰胺的合成(zec1)
[0225]
制备方法同化合物za1,以2
‑
氨基
‑5‑
甲氧基苯并噻唑和8b为原料,制得白色粉末状固体zec1,收率51%。
[0226]1h nmr(600mhz,dmso
‑
d6)δ12.67(s,1h),8.11(dd,j=8.5,2.0hz,1h),8.05(d,j=1.3hz,1h),7.90(d,j=8.7hz,1h),7.32(s,1h),7.28(d,j=8.7hz,1h),7.00(dd,j=8.7,2.3hz,1h),5.08(s,2h),3.87(s,3h),2.48(s,3h),2.29(s,3h),2.23(s,3h).
[0227]
《实施例49》n
‑
(苯并[d]噻唑
‑2‑
基)
‑4‑
((3,5
‑
二甲基异恶唑
‑4‑
基)甲氧基)
‑3‑
甲氧基苯甲酰胺的合成(zec3)
[0228]
制备方法同化合物za1,以2
‑
氨基苯并噻唑和8b为原料,制得白色粉末状固体zec3,收率65%。
[0229]1h nmr(600mhz,dmso
‑
d6)δ12.73(s,1h),8.12(dd,j=8.5,2.0hz,1h),8.06(s,1h),8.03(d,j=7.8hz,1h),7.80(d,j=8.0hz,1h),7.49(t,j=7.6hz,1h),7.36(t,j=7.5hz,1h),7.28(d,j=8.6hz,1h),5.09(s,2h),2.48(s,3h),2.29(s,3h),2.23(s,3h).
[0230]
《实施例50》4
‑
((3,5
‑
二甲基异恶唑
‑4‑
基)甲氧基)
‑3‑
甲基
‑
n
‑
(6
‑
(三氟甲氧基)苯并[d]噻唑
‑2‑
基)苯甲酰胺的合成(zec5)
[0231]
制备方法同化合物za1,以利鲁唑和8b为原料,制得白色粉末状固体zec5,收率63%。
[0232]1h nmr(600mhz,dmso
‑
d6)δ12.82(s,1h),8.16(d,j=1.7hz,1h),8.10(dd,j=8.5,2.2hz,1h),8.04(d,j=1.6hz,1h),7.86(d,j=8.7hz,1h),7.45(dd,j=8.6,1.9hz,1h),7.26(d,j=8.7hz,1h),5.07(s,2h),2.45(s,3h),2.26(s,3h),2.20(s,3h).
[0233]
《实施例51》4
‑
((3,5
‑
二甲基异恶唑
‑4‑
基)甲氧基)
‑3‑
甲基
‑
n
‑
(4
‑
甲基苯并[d]噻唑
‑2‑
基)苯甲酰胺的合成(zec6)
[0234]
制备方法同化合物za1,以2
‑
氨基
‑4‑
甲基苯并噻唑和8b为原料,制得白色粉末状固体zec6,收率77%。
[0235]1h nmr(600mhz,dmso
‑
d6)δ12.68(s,1h),8.11(dd,j=8.5,2.2hz,1h),8.05(d,j=1.6hz,1h),7.82(d,j=7.7hz,1h),7.25(dt,j=15.6,7.4hz,3h),5.06(s,2h),2.63(s,3h),2.45(s,3h),2.26(s,3h),2.20(s,3h).
[0236]
《实施例52》4
‑
((3,5
‑
二甲基异恶唑
‑4‑
基)甲氧基)
‑3‑
甲氧基
‑
n
‑
(6
‑
(三氟甲氧基)苯
‑
o[d]噻唑
‑2‑
基)苯甲酰胺的合成(zec7)
[0237]
制备方法同化合物za1,以利鲁唑和8a为原料,制得白色粉末状固体zec5,收率78%。
[0238]1h nmr(600mhz,dmso
‑
d6)δ12.92(s,1h),8.17(s,1h),7.91
–
7.82(m,2h),7.81(d,j
=1.8hz,1h),7.46(dd,j=8.7,1.6hz,1h),7.28(d,j=8.5hz,1h),5.03(s,2h),3.88(s,3h),2.42(s,3h),2.23(s,3h).
[0239]
《实施例53》4
‑
((3,5
‑
二甲基异恶唑
‑4‑
基)甲氧基)
‑3‑
甲氧基
‑
n
‑
(6
‑
(三氟甲氧基)苯
‑
o[d]噻唑
‑2‑
基)苯甲酰胺的合成(zal1)
[0240]
将3,5
‑
二甲基
‑4‑
羟甲基异噁唑溶于无水二氯甲烷,室温搅拌,于0℃氮气氛下滴加三溴化磷(pbr3 2.0eq)。由0℃升至室温搅拌4h后tlc检测反应完全,反应液由无色透明变成浅黄色,加入饱和碳酸氢钠溶液至ph为7
‑
8,二氯甲烷萃取,无水硫酸钠干燥,减压蒸发溶剂,残余物经柱层析分离得到无色透明有刺激性油状物。将4
‑
羟基苯乙酸酸(1.0eq)和氢氧化钾(koh 2.5eq)溶于乙醇:水为9:1的溶剂中,室温搅拌,向反应体系中滴加化合物上部反应得到的无色透明油状物,加热回流。12h后tlc检测反应完全。冷却至室温后,向反应体系中滴加6m hcl至溶液ph为2
‑
3,并伴有白色固体产生,减压抽滤,烘干,得到的白色固体即为化合物2
‑
(4
‑
((3,5
‑
二甲基异恶唑
‑4‑
基)甲氧基)苯基)乙酸,收率58%。
[0241]
制备方法同化合物za1,以2
‑
(4
‑
((3,5
‑
二甲基异恶唑
‑4‑
基)甲氧基)苯基)乙酸和4
‑
(4
‑
甲氧基苯基)噻唑
‑2‑
胺为原料,制得白色粉末状固体zal1,收率68%。
[0242]1h nmr(600mhz,dmso
‑
d6)δ12.41(s,1h),7.83(d,j=8.2hz,2h),7.44(s,1h),7.28(d,j=8.1hz,2h),6.98(t,j=8.2hz,4h),4.89(s,2h),3.79(s,3h),3.71(s,2h),2.39(s,3h),2.20(s,3h).
[0243]
《实施例54》n
‑
(苯并[d]噻唑
‑2‑
基)
‑2‑
(4
‑
((3,5
‑
二甲基异恶唑
‑4‑
基)甲氧基)苯基)乙酰胺的合成(zel1)
[0244]
制备方法同化合物za1,以2
‑
(4
‑
((3,5
‑
二甲基异恶唑
‑4‑
基)甲氧基)苯基)乙酸和2
‑
氨基苯并噻唑为原料,制得白色粉末状固体zel1,收率55%。
[0245]1h nmr(600mhz,dmso
‑
d6)δ12.56(s,1h),7.96(d,j=7.9hz,1h),7.75(d,j=8.0hz,1h),7.43(t,j=7.6hz,1h),7.30(t,j=7.9hz,3h),6.99(d,j=8.1hz,2h),4.89(s,2h),3.77(s,2h),2.39(s,3h),2.20(s,3h).
[0246]
《实施例55》2
‑
(4
‑
((3,5
‑
二甲基异恶唑
‑4‑
基)甲氧基)苯基)
‑
n
‑
(5
‑
甲氧基苯并[d]噻唑
‑2‑
基)乙酰胺的合成(zel2)
[0247]
制备方法同化合物za1,以2
‑
(4
‑
((3,5
‑
二甲基异恶唑
‑4‑
基)甲氧基)苯基)乙酸和2
‑
氨基
‑5‑
甲氧基苯并噻唑为原料,制得白色粉末状固体zel2,收率77%。
[0248]1h nmr(600mhz,dmso
‑
d6)δ12.52(s,1h),7.82(d,j=8.6hz,1h),7.29(d,j=8.3hz,3h),6.98(d,j=7.9hz,2h),6.93(d,j=8.7hz,1h),4.89(s,2h),3.82(s,3h),3.76(s,2h),2.39(s,3h),2.20(s,3h).
[0249]
《实施例55》苯甲酰胺衍生物对lncap细胞中ar的转录抑制活性数据。
[0250]
具体步骤是:将生长状态良好的lncap细胞用胰酶消化后,稀释成约1.2
×
105个/ml的细胞悬液,按照每孔500μl体积,接种于24孔板中,培养24h后,每孔转染100ng psa
‑
luc和2ng pcmv
‑
renilla质粒;24h后,将已调配好浓度的化合物溶液用rpmi 1640完全培养基稀释,吸弃24孔板中旧的培养基,加入给药后的新鲜培养基,同时设好阳性药对照组和空白组。24h后,吸弃原来的培养基,然后在每个孔中加入100μl1
×
plb裂解液,后将孔板防止在高速摇床震荡20min混匀,吸取细胞裂解液1.5ml于ep管中,12000rpm离心1min,取上清液20μl到白色不透明96孔板中,按照双荧光素酶报告系统检测试剂盒使用指南,使用960酶标仪
测定荧光值。
[0251]
本实施例主要提供了结构式(i)和(ii)化合物中的部分代表化合物对lncap细胞中ar的转录抑制活性数据(表1)。
[0252]
表1实施例化合物抗ar的转录抑制活性
[0253][0254]
《实施例56》苯甲酰胺衍生物对前列腺癌细胞lncap及pc
‑
3的细胞毒性。
[0255]
将生长状态良好的lncap和pc
‑
3细胞用胰蛋白酶消化后,稀释成约2
×
104个/ml细胞悬液,于96孔板的每个孔中接种200μl细胞悬液,培养24h后,将不同浓度梯度的药物溶液加入96孔板中,每孔加入1μl,不同浓度点药物分别做三组平行,同时设好空白组和阳性药对照组。继续培养72h后,分别向每孔中加入5g/l mtt的pbs溶液20μl,继续放置在二氧化碳孵箱中培养2
‑
4h,随后将96孔板从培养箱中取出,小心吸弃96孔板中的培养基,每孔加入100μl异丙醇使沉淀物溶解,将96孔板置于高速摇床上震荡混匀20min,使用多功能酶标仪在570nm波长下测定每孔吸光度值。以dmso组为100%进行归一化处理。从表2可以看出,化合物za1、za3和za5对于ar阳性的lncap细胞的抗增殖具有较高的活性,并且对于ar阴性的
pc
‑
3细胞的增殖影响较小,表明化合物具有较好的选择性。
[0256]
表2:实施例的前列腺癌细胞毒性研究
[0257][0258]
《实施例57》苯甲酰胺衍生物对ar蛋白下调的影响
[0259]
将生长状态良好的lncap、vcap与22rv1细胞用胰酶消化后,稀释成约2
×
105个/ml的细胞悬液,按照每孔2ml体积,接种于6孔板中,培养48h后,将已按照相应浓度配置好的药物溶液加入6孔板中,每孔加入8μl,同时设好空白组、激活组和阳性药对照组。继续培养24h后,收取蛋白样品。
[0260]
细胞蛋白样品收取:吸弃6孔板中培养基,每孔加入80μl预冷ripa(强)裂解液,刮取6孔板中细胞,收集到1.5ml ep管中。冰浴裂解30min,每隔5min震荡混匀一次。随后加入20μl 5
×
蛋白上样缓冲液裂解细胞。100℃金属浴煮30min。蛋白样品储存于
‑
20℃或者
‑
80℃。western blot流程:80v 40min,120v 100min电压下,使用已配制好的10%sds
‑
page分离蛋白样品。70v恒压湿法转膜80min(使用pvdf膜),用5%脱脂奶粉室温低速摇晃封闭1h。一抗孵育过夜,洗膜,二抗孵育1h,洗膜,然后ecl显色。抗体使用浓度为ar(1:500),psa(1:100),β
‑
actin(1:5000),山羊抗兔(1:5000),山羊抗小鼠(1:5000),兔抗山羊(1:5000)。从图8可以看出,化合物za5、za11、zc1均可浓度依赖地下调lncap细胞中的ar水平。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1