一种碘佛醇多取代物的合成的制作方法
1.本发明涉及一种碘佛醇多取代物的制备方法,属于化学合成技术领域。
背景技术:
2.碘佛醇(ivoersol)是美国万灵科医药有限公司(mallinckrodt medical inc.)研发的一种新型的非离子型造影剂,1988年被美国fda批准上市,目前已在日本、英国、法国、中国等地区上市。碘佛醇的化学名n,n
′‑
双(2,3二羟丙基)-5-[n-(2-羟乙基)-羟乙酰胺基]-2,4,6-三碘-1,3
‑ꢀ
苯二甲酰胺,化学结构式如式i所示:
[0003][0004]
目前文献报道碘佛醇合成工艺路线较多,主要路线有us4396598a报道了一种碘佛醇的制备方法,以5-氨基-2,4,6-三碘异酞酸为起始物料,经酰氯化、取代、乙酰基保护,以乙酰氧基乙酰氯为酰化试剂,先酰化后水解,再与氯乙醇反应制备碘佛醇。
[0005][0006]
专利us5648536报道了以5-氨基-n,n
’‑
双(2,3-二羟丙基)-2,4,6-三碘-苯二酰胺为起始原料,先与氯乙酰反应,再经过醋酸钠水解,最后与氯乙醇反应制备碘佛醇。
[0007][0008]
cn1477093a则以5-氯乙酰氨基-n,n
′‑
双-(2,3-二羟基丙基)-2,4,6-三碘-1,3-苯二甲酰胺和氯乙醇为原料,“一锅两步法”制备碘佛醇的方法。
[0009][0010]
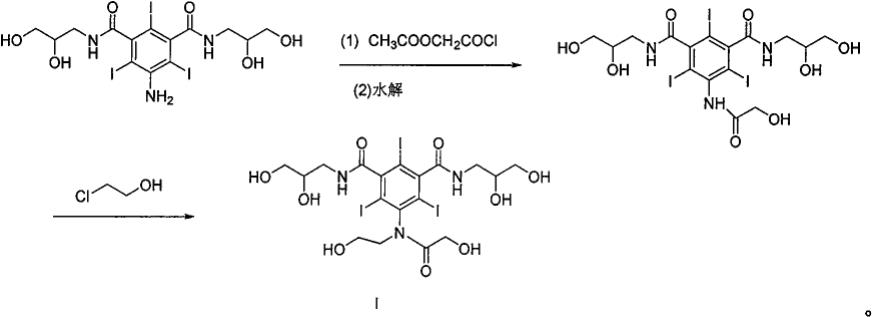
n,n
′‑
双(2,3二羟丙基)-5-[n-(2-羟乙基)-羟乙酰胺基]-2,4,6-三碘-1,3-苯二甲酰胺(碘佛醇),过量的氯乙醇同样在后续反应中碱性条件下可以生成碘佛醇多取代物(化学结构式如式 ii所示)。
[0011]
碘佛醇多取代物产生过程:
[0012][0013]
碘佛醇中多取代产物是不可避免的产生,合成碘佛醇粗品中含多取代物ii约1~2%,碘氟醇粗品母液中含量仅为4~5%。碘佛醇多取代产物进行质量研究需要纯度及含量高的对照品,因此需要对粗品母液进行多次柱层析纯化及液相制备,才能纯化出很少量碘佛醇多取代产物。为了便于控制反应条件避免产生碘佛醇多取代产物,了解多取代产物产生过程及该多取代产物质量研究,制备碘佛醇多取代产物尤为必要。通过定向合成手段可以高效大量制备碘佛醇多取代产物对照品,为碘氟醇原料药工艺和质量控制研究提供有力保障。为此,本发明在综合上述情况下,提供一种新的制备碘佛醇多取代物化合物(化学结构式如式ii所示) 的制备方法。
技术实现要素:
[0014]
本发明所要解决的技术问题是针对现有技术中制备及纯化困难的不足,而提供一种碘佛醇多取代物的制备方法,本发明方法中,碘佛醇多取代产物(ii)的收率>70%、纯度和含量均大于99%,制备的碘佛醇多取代产物(ii)可以用于碘佛醇工艺及质量研究。
[0015]
为了实现上述目的,本发明采用如下技术方案:
[0016]
一种碘佛醇多取代物的制备方法,包括以下步骤:
[0017]
(1)以式iii所示的5-氨基-n,n
′‑
双-(2,3-二羟基丙基)-2,4,6-三碘-1,3-苯二甲酰胺为原料,在极性溶剂i中,与2-(2-氯乙氧基)乙酰氯进行缩合反应,得到式iv所示的中间化合物,化学反应式如下所示:
[0018][0019]
(2)式iv所示的化合物在极性溶剂ii中,在碱存在的条件下与氯乙醇进行取代反应,取代反应结束后;利用2mol/l稀盐酸将体系ph调节为7.5~8.5,然后向体系中加入乙酸钠和纯化水,加热回流进行水解反应,水解反应结束后得到式ii所示的碘佛醇多取代物,化学反应式如下所示:
[0020][0021]
上述技术方案中,步骤(1)中,所述的5-氨基-n,n
′‑
双-(2,3-二羟基丙基)-2,4,6-三碘-1,3
‑ꢀ
苯二甲酰胺和2-(2-氯乙氧基)乙酰氯,摩尔比为1∶(0.8~1.2),摩尔比优选为1∶(0.9~1.1)。
[0022]
上述技术方案中,步骤(1)中,所述的极性溶剂i为n,n-二甲基甲酰胺、n,n-二甲基乙酰胺、1,4-二氧六环中的任意一种、两种及以上以任意比例混合而成的混合物,优选为 n,n-二甲基乙酰胺。
[0023]
上述技术方案中,步骤(1)中,所述的极性溶剂i与5-氨基-n,n
′‑
双-(2,3-二羟基丙基)-2,4,6-三碘-1,3-苯二甲酰胺(iii),质量比为1∶(1~4),质量比优选为1∶(2~3)。
[0024]
上述技术方案中,步骤(1)中,所述的缩合反应,反应温度为-10~10℃,反应时间为2~6h,优选为反应温度0~5℃、反应时间为3~5h。
[0025]
上述技术方案中,步骤(1)中,所述的缩合反应结束后,需要采取后处理:向体系中加入0.5%盐酸水溶液搅拌析晶,经过滤、洗涤、干燥后得到式iv所示的化合物。
[0026]
上述技术方案中,步骤(2)中,所述的式iv所示化合物与氯乙醇,摩尔比为1∶(2.5~3.5),优选为1∶(2.8~3.2)。
[0027]
上述技术方案中,步骤(2)中,所述的极性溶剂ii为水、甲醇、乙醇、异丙醇中的任
意一种、两种及以上以任意比例混合而成的混合物,优选为水和甲醇中的任意一种或两种上以任意比例混合而成的混合物。
[0028]
上述技术方案中,步骤(2)中,所述的极性溶剂ii与式iv所示化合物,质量比为1∶ (1~4)。质量比优选为1∶(2~3)。
[0029]
上述技术方案中,步骤(2)中,所述的碱为氢氧化钠、氢氧化钾、氢氧化锂、碳酸铯中的任意一种、两种及以上以任意比例混合而成的混合物,优选为氢氧化钠和氢氧化钾中的任意一种或两种上以任意比例混合而成的混合物。
[0030]
上述技术方案中,步骤(2)中,所述的式iv所示化合物与碱,摩尔比为1∶(1.5~2.5),摩尔比优选为1∶(1.8~2.2)。
[0031]
上述技术方案中,步骤(2)中,所述的取代反应,反应条件为:反应温度为35~75℃,反应时间为8~13;优选为:反应温度55~65℃,反应时间为10~12h。
[0032]
上述技术方案中,步骤(2)中,所述的水解反应中,乙酸钠与式iv所示化合物的质量比为(0.5~4)∶1,质量比优选为(1.5~3)∶1。
[0033]
上述技术方案中,步骤(2)中,所述的水解反应中,加入的纯化水与式iv所示化合物的质量比为(2~5)∶1,质量比优选为(3~4)∶1。
[0034]
上述技术方案中,步骤(2)中,所述的水解反应,加热回流反应时间为18~26h,优选为20~24h。
[0035]
上述技术方案中,步骤(2)中,所述的水解反应结束后,降温至室温后进行减压浓缩;浓缩产物采用xad-16大孔吸附树脂进行纯化,洗脱剂为纯化水;纯化后得到的产物加入甲醇进行析晶,经过滤、干燥后得到式ii所示的碘佛醇多取代物。
[0036]
本发明方法,通过可商业化物料通过2步常规反应制备纯度大于等于99%的碘佛醇多取代物(结构式如ii所示),两步收率可达到70%以上,通过标定可用于碘佛醇的质量研究。
具体实施方式
[0037]
以下对本发明技术方案的具体实施方式详细描述,但本发明并不限于以下描述内容:
[0038]
本发明所述含量或者纯度通过hplc面积归一化法检测数据。
[0039]
下面结合具体的实施例,对本发明进行阐述;
[0040]
实施例1
[0041]
一种碘佛醇多取代物的制备方法,包括以下步骤:
[0042]
(1)100ml反应瓶中加入式iii所示的化合物(7.05g,10mmol)、n,n-二甲基乙酰胺14ml,搅拌降温至0~5℃。滴加2-(2-氯乙氧基)乙酰氯(1.39g n,n-二甲基乙酰胺,10mmol),滴加过程控温10℃以下。滴加完毕,保温0~5℃搅拌反应4h,tlc监控反应结束。反应毕,反应液中加入预冷0.5%盐酸水溶液60ml(10
±
5℃),析出固体后,10~20℃搅拌3h,过滤,滤饼加1体积纯化水洗涤,收集滤饼,50
±
5℃鼓风干燥至8h,得到类白色固体7.25g(式iv所示),纯度:98.2%,收率90%。
[0043]
(2)称取纯化水20ml(作为极性溶剂ii使用)加入到100ml反应瓶中,搅拌,称取 naoh(0.88g,15.6mmol),加入到反应体系中。称取式iv所示的化合物(7.00g,8.67mmol) 加
入到反应体系中,加热升温55-60℃搅拌至原料溶解,加入氯乙醇(2.10g,26.01mmol),保温55
±
5℃反应10h,降温至室温,用2mol/l稀盐酸调节ph7.5~8.5,加入乙酸钠10.5g和纯化水21g,继续加热升温至100℃回流反应20h,hplc检测反应结束,降温至室温,50~60℃下减压浓缩至干。
[0044]
大孔吸附树脂直径3厘米,xad-16树脂填装高50厘米,纯化水洗脱活化。
[0045]
将残留物溶解于10ml纯化水中,上样,纯化水洗脱,先将盐洗脱出来,硝酸银水溶液检测无浑浊。再用约15-20柱体积超纯水洗脱出产品,收集产品。50~60℃减压浓缩干,加入甲醇15ml析出大量固体,室温搅拌3h过滤,滤饼50~60℃干燥得到多取代物ii 6.02g,纯度 99.5%,收率:82%,滴定含量:99.2%。
[0046]
实施例2
[0047]
一种碘佛醇多取代物的制备方法,包括以下步骤:
[0048]
(1)100ml反应瓶中加入式iii所示的化合物(14.1g,20mmol)、n,n-二甲基甲酰胺30ml,搅拌降温至0~5℃。滴加2-(2-氯乙氧基)乙酰氯(3.06g,22mmol),滴加过程控温10℃以下。滴加完毕,保温0~5℃搅拌反应4h,tlc监控反应结束。反应毕,反应液中加入预冷0.5%盐酸水溶液120ml(10
±
5℃2),析出固体后,10~20℃搅拌3h,过滤,滤饼加1体积纯化水洗涤,收集滤饼,50
±
5℃鼓风干燥至8h,得到类白色固体14.12g(式iv所示),纯度:98.1%,收率87.6%
[0049]
(2称取甲醇35ml加入到100ml反应瓶中,搅拌,称取naoh(1.42g,35.44mmol),加入到反应体系中。称取化合物2(13.00g,16.11mmol)加入到反应体系中,加热升温55-60℃搅拌至原料溶解,加入氯乙醇(4.15g,51.54mmol),保温55
±
5℃反应10h,降温至室温,用 2mol/l稀盐酸调节ph7.5~8.5,加入乙酸钠26g和纯化水39g,继续加热升温70~80℃至回流反应24h,hplc检测反应结束,降温至室温,50~60℃下减压浓缩干。
[0050]
大孔吸附树脂直径5厘米,xad-16树脂填装高50厘米,纯化水洗脱活化。
[0051]
将残留物溶解于15ml纯化水中,上样,纯化水洗脱,先将盐洗脱出来,硝酸银水溶液检测无浑浊。再用约15-20柱体积超纯水洗脱出产品,收集产品。50~60℃减压浓缩干,加入甲醇25ml析出大量固体,室温搅拌3h过滤,滤饼50~60℃干燥得到多取代物ii 11.24g,纯度 99.1%,收率:82%,滴定含量:99.0%。
[0052]
上述实例只是为说明本发明的技术构思以及技术特点,并不能以此限制本发明的保护范围。凡根据本发明的实质所做的等效变换或修饰,都应该涵盖在本发明的保护范围之内。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1