环丙烷骨架双膦配体与其钴配合物和制备方法及应用
1.本发明涉及环丙烷骨架双膦配体与其钴配合物和制备方法及其应用。具体的讲是以双芳基重氮与富马酸酯进行环丙烷化反应制备反式取代的环丙烷-1,2-二酯,经后续的还原、取代等反应得到具有偕二芳基取代的反式环丙烷双膦配体,将其与二氯化钴进行络合反应,可以制备相应的环丙烷双膦配体钴的配合物。在活化试剂存在下,该双膦配体钴配合物能够催化内炔烃与频哪醇硼烷的硼氢化反应,得到烯基硼化合物,表现出很高的活性和独特的选择性,具有很好的应用前景。
背景技术:
2.有机硼化合物是分子内含有c-b键化合物的统称,是一类重要的功能分子,被广泛应用于医药,材料及有机合成等领域[(1)hall,d.g.boronic acids:preparation,applications in organic synthesis and medicine;john wiley & sons:weinheim,2006.(2)coca,a.boron reagents in synthesis;acs symposium series;american chemical society:washington,dc,2016.],因此有机硼化合物的合成与应用研究具有重要意义。经典合成有机硼化合物的方法包括锂试剂或格氏试剂对双硼试剂的加成以及过渡金属催化的miyaura硼化反应,尽管现有方法应用广泛,但存在着原料需预官能团化,反应官能团兼容性差,反应原子经济性差等问题,因此发展高效的有机硼化合物制备方法具有重要意义。因原料来源广泛,反应条件温和,操作简单,近年来不饱和键的催化硼氢化反应成为研究热点[(1)obligacion,j.v.;chirik,p.j.nat.rev.chem.2018,2,15.(2)yoshida,h.acs catal.2016,6,1799]。
[0003]
相对于端炔,内炔的硼氢化反应活性通常较低,存在化学选择性(硼氢化或氢化)问题;当内炔两端取代基位阻或电性差异不大时,区域选择性也难以控制;由于异构化等过程的存在,内炔硼氢化反应还可能存在立体选择性(顺式加成或反式加成)问题。总之,内炔硼氢化反应还存在化学、区域和立体选择性难以调控的问题。
[0004]
从底物类型划分,芳基烷基内炔的硼氢化反应已有较多报道,但仍有一些问题尚未解决。其中,芳基烷基内炔硼氢化的顺式α-加成(烯基硼产物中硼与氢加成在双键的同一侧,且硼加成至靠近芳基一端)选择性已经有几例报道,可以通过铜搭配双膦配体或氮杂环卡宾配体来实现[(1)semba,k.;fujihara,t.;terao,j.;tsuji,y.chem.eur.j.2012,18,4179.(2)bidal,y.d.;lazreg,f.;cazin,c.s.j.acs catal.2014,4,1564.(3)hall,j.w.;unson,d.m.l.;brunel,p.;collins,l.r.;cybulski,m.k.;mahon,m.f.;whittlesey,m.k.organometallics 2018,37,3102]。然而,芳基烷基内炔硼氢化的顺式β-加成(烯基硼产物中硼与氢加成在双键的同一侧,且硼加成至靠近烷基一端)选择性则并无系统研究,目前在路易斯酸,如三芳基硼催化下可以实现该转化,但反应的区域选择性依赖于内炔底物两端取代基的位阻差异[(1)fleige,m.;j.;vom stein,t.;glorius,f.;stephan,d.w.chem.commun.2016,52,10830.(2)lawson,j.r.;wilkins,l.c.;melen,r.l.chem.eur.j.2017,23,10997.(3)bismuto,a.;cowley,m.j.;thomas,
s.p.adv.synth.catal.2021,363,2382]。除此之外,使用大位阻的硼氢化试剂,也有助于内炔硼氢化反应的区域选择性控制。2009年,suginome,m.等人[iwadate,n.;suginome,m.org.lett.2009,11,1899]报道了铱催化的以hbdan为硼氢化试剂的内炔硼氢化反应,实现了1-苯基-1-丙炔硼氢化的顺式β-加成选择性控制,但所用硼氢化试剂不易得且底物适用性不明。
[0005]
对于双烷基内炔的区域和立体选择性硼氢化反应,主要依靠底物控制(位阻差异或导向作用)。其中,双烷基内炔硼氢化的顺式β-加成(烯基硼产物中硼与氢加成在双键的同一侧,且硼加成至小位阻烷基一端)选择性只有零星的报道,硼试剂[nieto-sepulveda,e.;bage,a.d.;evans,l.a.;hunt,t.a.;leach,a.g.;thomas,s.p.;lloyd-jones,g.c.j.am.chem.soc.2019,141,18600.]和铝试剂[bismuto,a.;thomas,s.p.;cowley,m.j.angew.chem.int.ed.2016,55,15356.]都可以催化双烷基内炔的硼氢化反应,硼倾向于加成至小位阻的烷基一端,但反应的区域选择性取决于内炔底物两端取代基位阻的差异。而双烷基内炔硼氢化的顺式α-加成(烯基硼产物中硼与氢加成在双键的同一侧,且硼加成至大位阻烷基一端)选择性目前并无有效的催化体系,仅在一例铜催化的炔丙醚衍生物的硼氢化反应中有所体现[semba,k.;fujihara,t.;terao,j.;tsuji,y.chem.eur.j.2012,18,4179.],此处底物中的醚官能团可能起到了关键性的导向作用。
[0006]
总而言之,对于内炔硼氢化反应,通常为顺式加成,硼倾向于加成到小位阻一端,但反应的区域选择性很难实现自由调控。对于芳基烷基内炔的硼氢化反应,顺式α-加成选择性已经得到了良好解决(铜氢体系),该类底物的顺式β-加成选择性还需进一步提高。对于双烷基内炔的硼氢化反应,其加成的选择性尚无通用的控制手段。因此发展用于内炔硼氢化反应的新型过渡金属催化剂,特别是基于丰产金属的催化剂,克服已知催化剂存在的缺点,实现文献中还不能很好控制的区域选择性,是本领域研究的重点之一。
技术实现要素:
[0007]
本发明的目的在于提供一种环丙烷骨架双膦配体与其钴配合物的制备方法及其应用,以克服已有技术的不足。
[0008]
本发明所述的环丙烷骨架双膦配体(i),其特征在于具有如下的结构式:
[0009][0010]
其中:
[0011]
r1、r2为苯基、取代的苯基,r3、r4为苯基、取代的苯基、c
1-c8烷基,r1、r2、r3、r4可以相同,也可以不同;
[0012]
所述取代的苯基,取代基为c
1-c8烷基、c
1-c8烷氧基、c
2-c8酰氧基、羟基、卤素、氨基、(c
1-c8酰基)氨基、二(c
1-c8烷基)氨基、c
1-c8酰基、c
2-c8酯基、卤代烷中的一种或几种;取代基数目为0-5;
[0013]
所述的环丙烷骨架双膦配体(i),其特征在于:
[0014]
所述的c
1-c8烷基为甲基、乙基、正丙基、异丙基、环丙基、正丁基、异丁基、仲丁基、叔丁基、正戊基、异戊基、新戊基、仲戊基、叔戊基、正己基、异己基、新己基、仲己基、叔己基、正庚基、异庚基、新庚基、仲庚基、叔庚基、正辛基、异辛基、新辛基、仲辛基或叔辛基;
[0015]
所述的c
1-c8酰基为甲酰基、乙酰基、丙酰基、正丁酰基、异丁酰基、正戊酰基、异戊酰基、仲戊酰基、新戊酰基、正己酰基、异己酰基、新己酰基、仲己酰基、正庚酰基、异庚酰基、新庚酰基、仲庚酰基、正辛酰基、异辛酰基、新辛酰基、仲辛酰基、1-环丙基甲酰基、1-环丁基甲酰基、1-环戊基甲酰基、1-环己基甲酰基、1-环庚基甲酰基;
[0016]
所述的c
2-c8酰氧基为乙酰氧基、丙酰氧基、正丁酰氧基、异丁酰氧基、正戊酰氧基、异戊酰氧基、仲戊酰氧基、新戊酰氧基、正己酰氧基、异己酰氧基、新己酰氧基、仲己酰氧基、正庚酰氧基、异庚酰氧基、新庚酰氧基、仲庚酰氧基、正辛酰氧基、异辛酰氧基、新辛酰氧基、仲辛酰氧基、1-环丙基甲酰氧基、1-环丁基甲酰氧基、1-环戊基甲酰氧基、1-环己基甲酰氧基、1-环庚基甲酰氧基;
[0017]
所述的c
2-c8酯基为甲氧羰基、乙氧羰基、丙氧羰基、异丙氧羰基、丁氧羰基、异丁氧羰基、正戊氧羰基、异戊氧羰基、新戊氧羰基、仲戊氧羰基、叔戊氧羰基、环戊氧羰基、正己氧羰基、异己氧羰基、新己氧羰基、仲己氧羰基、叔己氧羰基、环己氧羰基、正庚氧羰基、异庚氧羰基、新庚氧羰基、仲庚氧羰基、叔庚氧羰基、环庚氧羰基;
[0018]
所述的卤代烷基为含氟、氯、溴或碘的卤代烷基。
[0019]
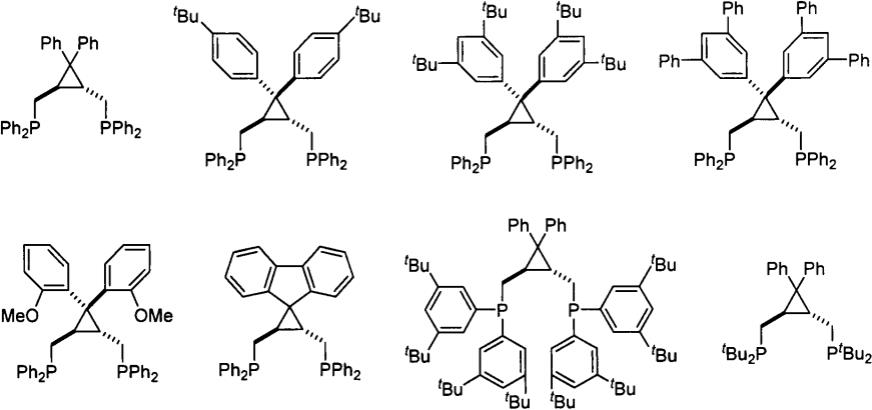
所述的环丙烷骨架双膦配体,其特征在于它是:
[0020][0021]
这些双膦配体可以是外消旋体、左旋体和右旋体。
[0022]
所述的消旋环丙烷骨架双膦配体的制备方法,其特征在于它是经过如下步骤制备:
[0023]
(1)在苯、甲苯、四氢呋喃、乙酸乙酯、乙腈中的一种或几种溶剂中,50-120℃下,富马酸二甲酯与重氮化合物进行环丙烷化,反应1-24小时,制备得到反式酯基取代的环丙烷骨架中间体,其反应式为:
[0024][0025]
(2)在甲苯、乙醚、叔丁基甲基醚、四氢呋喃中的一种或几种溶剂中,-30-30℃下,
以四氢铝锂为还原剂,将(1)产物中酯基还原为羟亚甲基,反应1-24小时,制备得到反式羟亚甲基取代的环丙烷骨架中间体,其反应式为:
[0026][0027]
(3)在二氯甲烷、氯仿、四氢呋喃中的一种或几种溶剂中,-30-30℃下,以三乙胺为碱,将(2)制得的环丙烷二醇中间体与甲磺酰氯反应,反应1-24小时,制备得到具有甲磺酸酯离去基团的环丙烷骨架中间体,其反应式为:
[0028][0029]
(4)在四氢呋喃、乙醚、叔丁基甲基醚中的一种或几种溶剂中,-78-30℃下,以正丁基锂为碱,先与二取代膦氢作用,生成二取代膦锂试剂,再加入(3)中产物,令二取代膦基取代甲磺酸酯,反应1-24小时,制备得到反式取代的环丙烷骨架双膦配体,其反应式为:
[0030][0031]
其中,r
1-r4如化合物(i)所定义。
[0032]
所述的光学环丙烷骨架双膦配体的制备方法,其特征在于它是经过如下步骤制备:
[0033]
(a)在二氯甲烷、氯仿、甲苯、四氢呋喃中的一种或几种溶剂中,-30-30℃下,以三乙胺为碱,将富马酰氯与光学的联萘酚单甲醚进行缩合,反应1-24小时,制备得到含有手性辅基的富马酸酯中间体,其反应式为:
[0034][0035]
(b)首先,在苯、甲苯、四氢呋喃、乙酸乙酯、乙腈中的一种或几种溶剂中,50-120℃下,将(a)制备的光学的富马酸酯与二苯基重氮进行环丙烷化反应,反应1-24小时,制备得到一对可以分离提纯的非对映异构体。接下来,在甲苯、乙醚、叔丁基甲基醚、四氢呋喃中的一种或几种溶剂中,-30-30℃下,以四氢铝锂为还原剂,将上述分离的非对映异构体分别进行还原,反应1-24小时,制备得到含反式羟亚甲基结构的环丙烷骨架中间体,它们是一对对映异构体。在此过程中,绝大部分光学的联萘酚单甲醚可以回收。其反应式为:
[0036][0037]
(c)在二氯甲烷、氯仿、四氢呋喃中的一种或几种溶剂中,-30-30℃下,以三乙胺为碱,将(b)制得的光学纯环丙烷二醇中间体与甲磺酰氯反应,反应1-24小时,制备得到具有甲磺酸酯离去基团的环丙烷骨架中间体,其反应式为:
[0038][0039]
(d)在四氢呋喃、乙醚、叔丁基甲基醚中的一种或几种溶剂中,-78-30℃下,以正丁基锂为碱,先与二取代膦氢作用,生成二取代膦锂试剂,再加入(c)制备的光学纯双甲磺酸酯中间体,令二取代膦基取代甲磺酸酯,反应1-24小时,制备得到反式取代的环丙烷骨架手性双膦配体,其反应式为:
[0040][0041]
其中,r
3-r4如化合物(i)所定义。
[0042]
所述的环丙烷骨架双膦配体钴配合物(ii),其特征在于具有如下的结构式:
[0043][0044]
其中:r
1-r4如化合物(i)所定义。
[0045]
所述的环丙烷骨架双膦配体钴配合物,其特征在于它是:
[0046][0047]
这些双膦配体钴配合物可以是外消旋体、左旋体和右旋体。
[0048]
所述的环丙烷骨架双膦配体钴配合物的制备方法,其特征在于它是经过如下步骤制备:在有机溶剂中,0-120℃下,环丙烷骨架双膦配体与二氯化钴络合1-60小时,制备得到环丙烷骨架双膦配体钴配合物,其反应式为:
[0049][0050]
其中,r
1-r4如化合物(i)所定义。络合反应所用有机溶剂为苯、甲苯、乙醚、叔丁基甲基醚、四氢呋喃、乙腈、二氯甲烷或氯仿中的一种或几种。
[0051]
所述的环丙烷骨架双膦配体钴配合物(ii)的应用,其特征在于它作为催化剂用于内炔的硼氢化反应:
[0052][0053]
其中:r
5-r6是苯基、取代苯基、杂芳基、烯基、烷基及官能团取代的烷基,hbpin是频哪醇硼烷。
[0054]
所述的环丙烷骨架双膦配体钴配合物的应用,其特征在于将催化剂和活化试剂加入反应管中,之后依次加入溶剂、频哪醇硼烷和内炔底物,在搅拌条件下反应至结束。
[0055]
所述的环丙烷骨架双膦配体钴配合物的应用,其特征在于所述的硼氢化反应条件是:催化剂用量为0.1-5mol%;活化试剂为有机锂、有机镁、有机铝、有机锌试剂、甲醇、乙醇、叔丁醇、碱金属盐(锂、钠、钾)、四(3,5-二(三氟甲基)苯基)硼酸钠(nabarf)中的一种或几种;所用溶剂是c
1-c8的醚类,甲苯,烷烃或底物本身(无溶剂条件)中的一种或几种;如有溶剂,底物浓度为0.1-10m;反应温度为0-120℃;反应时间为1-24小时。
[0056]
本发明的优点和有益效果:
[0057]
总而言之,将富马酸酯与双芳基重氮进行环丙烷化反应,得到的环丙烷二酯中间体经过后续的还原、取代等反应可以制备具有偕二芳基取代的反式环丙烷双膦配体,将得到的配体与与二氯化钴进行络合反应,可以制备相应的环丙烷骨架双膦配体钴配合物。该新型环丙烷骨架双膦配体钴配合物可以高效催化内炔的硼氢化反应,并表现出以下特点:在芳基烷基内炔的硼氢化反应中以当前最优的选择性实现了β-加成选择性控制,在双烷基内炔中实现了当前无法实现的α-加成选择性控制。除此之外,环丙烷骨架双膦配体钴配合物催化的内炔硼氢化反应还具有底物适用范围广,官能团耐受性好,选择性可调控等优点。上述特点表明,本发明所提供的环丙烷骨架双膦配体钴配合物克服了已有技术的缺点,是目前催化内炔硼氢化反应最为高效的催化剂之一,具有很好的应用前景。
附图说明
[0058]
图1为环丙烷骨架双膦配体(1s,1s)-1a的单晶结构;
[0059]
图2为环丙烷骨架双膦配体(1s,1s)-1i的单晶结构;
[0060]
图3为环丙烷骨架双膦配体钴配合物2a的单晶结构。
具体实施方式
[0061]
通过下述实施实例将有助于进一步理解本发明,但不应将此理解为本发明上述主题的范围仅限于以下的实施例,凡基于本发明上述内容所实现的技术均属于本发明的范围。
[0062]
一般说明:
[0063]
以下实例中使用了缩写,其含义如下:
[0064]
me是甲基,et是乙基,ipr是异丙基,
t
bu是叔丁基,ph是苯基,thf是四氢呋喃,dcm是二氯甲烷,dme是乙二醇二甲醚,mtbe是叔丁基甲基醚,pe是石油醚,ea是乙酸乙酯,mscl是甲基磺酰氯,ar是氩气,cdcl3是氘代氯仿,dibal-h是二异丁基氢化铝。
[0065]
equiv是当量,rt代表室温,s/c是底物与催化剂的物质的量之比,nd代表未检测到,tlc是薄层色谱,nmr是核磁共振,hrms是高分辨质谱,hplc是高效液相色谱,ir是红外吸收光谱,gc-ms是气相色谱-质谱联用。
[0066]
所用溶剂在使用前经标准操作提纯,干燥;所用试剂均为市售或按照已有文献方法合成得到,并在使用前提纯。
[0067]
实施例1:3,3-二芳基环丙烷-1,2-二羧酸二甲酯3a-3f的制备
[0068]
3,3-二苯基环丙烷-1,2-二羧酸二甲酯3a的合成:
[0069][0070]
在250ml圆底烧瓶中依次加入富马酸二甲酯(7.2g,50mmol)、甲苯(100ml)、二苯基重氮(11.5g,60mmol),圆底瓶上方装备回流冷凝管,通入冷凝水。之后将其置于250ml加热模块中,加热至110℃。随着反应的进行,可以明显观察到溶液颜色逐渐变浅,由紫色变为浅黄色。tlc监测反应,使用高锰酸钾进行显色。反应结束后,通过旋蒸除去溶剂甲苯,得到粗
产物进行硅胶柱层析分离(洗脱剂为石油醚/二氯甲烷=1∶1,v/v),得到化合物3a,熔点171-172℃。
[0071]1h nmr(400mhz,cdcl3)δ7.36-7.34(m,4h),7.27-7.23(m,4h),7.19-7.15(m,2h),3.50(s,6h),3.29(s,2h).
[0072]
13
c nmr(101mhz,cdcl3)δ169.08,140.05,128.57,128.51,127.27,51.98,46.99,32.08.
[0073]
核磁谱图数据与文献报道一致[walser,p.;renold,p.;n’goka,v.;hosseinzadeh,f.;tamm,c.helv.chim.acta 1991,74,1941]。
[0074]
以下化合物(3b-3f)的合成方法与实施例1相同,并且获得的环丙烷二酯中间体未经提纯,直接用于下一步还原反应。
[0075][0076]
实施例2:3,3-二芳基环丙烷-1,2-二亚甲基醇4a-4f的制备
[0077]
3,3-二苯基环丙烷-1,2-二亚甲基醇4a的合成:
[0078][0079]
在500ml圆底烧瓶中,加入3a和四氢呋喃(100ml),搅拌均匀。在0℃及搅拌条件下,向体系中滴加四氢呋喃(50ml)和lialh4(3.8g,100mmol)的混合物。滴加完毕,移去冷浴,室温搅拌反应2h。之后在0℃及搅拌条件下,向体系中缓慢滴加酒石酸钠钾的水溶液,以淬灭反应。淬灭完成后,向反应体系中加水(80ml),用分液漏斗分液,水相用mtbe萃取(100ml
×
3),合并有机相,用饱和食盐水洗涤有机相,之后用无水硫酸镁干燥。过滤,通过旋蒸浓缩滤液,所得粗产物以硅胶柱层析分离(洗脱剂为二氯甲烷/甲醇=20∶1,v/v)。之后用乙腈对产物进行重结晶。最终得到化合物4a 8.5g,为白色固体,两步反应总收率为67%,熔点为162-163℃。
[0080]1h nmr(400mhz,cdcl3)δ7.33-7.30(m,4h),7.29-7.24(m,4h),7.20-7.16(m,2h),3.44-3.42(m,4h),1.99-1.94(m,2h),1.53(br,2h).
[0081]
13
c nmr(101mhz,cdcl3)δ141.97,129.47,128.57,126.64,63.23,40.95,29.45.
[0082]
hrms(esi)calcd for[m+na,c
17h18
nao2]
+
:277.11990,found:277.11986.
[0083]
以下化合物的合成方法与实施例2相同
[0084]
3,3-二(4-叔丁基)苯基环丙烷-1,2-二亚甲基醇(4b)
[0085][0086]
白色固体,收率82%,熔点184-186℃。
[0087]1h nmr(400mhz,cdcl3)δ7.27-7.20(m,8h),3.58-3.49(m,2h),3.33-3.24(m,2h),2.18(br,2h),1.94-1.85(m,2h),1.26(s,18h).
[0088]
13
c nmr(101mhz,cdcl3)δ149.25,138.99,129.01,125.39,63.26,40.18,34.36,31.32,29.43.
[0089]
hrms(esi)calcd for[m+na,c
25h34
nao2]
+
:389.2451,found:389.2455.
[0090]
3,3-二(3,5-二叔丁基)苯基环丙烷-1,2-二亚甲基醇(4c)
[0091][0092]
白色固体,收率89%,熔点183-185℃。
[0093]1h nmr(400mhz,cdcl3)δ7.19(t,j=1.8hz,2h),7.16(d,j=1.8hz,4h),3.64(dd,j=11.5,5.8hz,2h),3.33(dd,j=11.4,6.3hz,2h),1.89-1.83(m,2h),1.75(br,2h),1.27(s,36h).
[0094]
13
c nmr(101mhz,cdcl3)δ150.56,141.50,123.48,120.04,63.79,42.15,34.73,31.44,30.11.
[0095]
hrms(esi)calcd for[m+na,c
33h50
nao2]
+
:501.3703,found:501.3705.
[0096]
3,3-二(3,5-二苯基)苯基环丙烷-1,2-二亚甲基醇(4d)
[0097][0098]
白色固体,收率50%,熔点121-129℃。
[0099]1h nmr(400mhz,cdcl3)δ7.63-7.56(m,14h),7.45-7.39(m,8h),7.36-7.31(m,4h),3.74(dd,j=11.5,5.6hz,2h),3.37(dd,j=11.5,6.7hz,2h),2.46(br,2h),2.17-2.06(m,2h).
[0100]
13
c nmr(101mhz,cdcl3)δ142.85,142.10,140.88,128.78,127.51,127.31,124.77,63.41,41.34,29.79.
[0101]
hrms(esi)calcd for[m+na,c
41h34
nao2]
+
:581.2451,found:581.2455.
[0102]
3,3-二(2-甲氧基)苯基环丙烷-1,2-二亚甲基醇(4e)
[0103][0104]
黄色固体,收率46%,熔点101-103℃。
[0105]1h nmr(400mhz,cdcl3)δ7.59(dd,j=7.6,1.8hz,2h),7.19(td,j=7.8,1.7hz,2h),6.98(td,j=7.5,1.1hz,2h),6.80(d,j=8.2hz,2h),3.83-3.77(m,2h),3.68(s,6h),3.11(d,j=6.4hz,2h),2.86(t,j=10.5hz,2h),1.80(ddd,j=9.6,6.8,4.8hz,2h).
[0106]
13
c nmr(101mhz,cdcl3)δ157.76,133.47,129.15,128.13,120.92,111.75,63.16,55.52,35.21,28.93.
[0107]
hrms(esi)calcd for[m+na,c
19h22
nao4]
+
:337.14103,found:337.14061.
[0108]
1-螺芴基环丙烷-2,3-二亚甲基醇(4f)
[0109][0110]
白色固体,收率59%,熔点136-138℃。
[0111]1h nmr(400mhz,cdcl3)δ7.81(d,j=7.5hz,2h),7.35(t,j=7.4hz,2h),7.26(td,j=7.6,1.3hz,2h),7.17(d,j=7.6hz,2h),3.98(dd,j=12.1,6.2hz,2h),3.90(dd,j=12.2,5.5hz,2h),3.09(br,2h),2.35-2.26(m,2h).
[0112]
13
c nmr(101mhz,cdcl3)δ144.13,140.34,126.74,126.41,121.46,120.09,60.59,37.95,37.43.
[0113]
hrms(esi)calcd for[m+na,c
17h16
nao2]
+
:275.1043,found:275.1048.
[0114]
实施例3:3,3-二芳基环丙烷-1,2-二亚甲基甲磺酸酯5a-5f的制备
[0115]
3,3-二苯基环丙烷-1,2-二亚甲基甲磺酸酯5a的合成:
[0116][0117]
于250ml圆底烧瓶中加入4a(1.96g,7.7mmol),二氯甲烷(50ml),三乙胺(3.2ml,23.1mmol,3equiv),搅拌均匀后,向体系中滴加溶于二氯甲烷(10ml)的甲磺酰氯(1.5ml,19.3mmol,2.5equiv)。加料完毕后,室温搅拌反应,tlc监测反应。4h后原料转化完全,停止搅拌,向体系中加入硅胶制砂,通过硅胶柱层析提纯产物(洗脱剂为pe/ea=1∶1,v/v)。获得无色油状产物2.50g,收率79%,分解温度105℃。
[0118]1h nmr(400mhz,cdcl3)δ7.34-7.26(m,8h),7.25-7.19(m,2h),4.16-4.08(m,2h),3.92-3.85(m,2h),2.85(s,6h),2.25-2.17(m,2h).
[0119]
13
c nmr(101mhz,cdcl3)δ139.74,129.53,128.78,127.40,69.93,42.43,37.26,
25.95.
[0120]
hrms(esi)calcd for[m+na,c
19h22
nao6s2]
+
:433.0750,found:433.0745.
[0121]
以下化合物的合成方法与实施例3相同
[0122]
3,3-二(4-叔丁基)苯基环丙烷-1,2-二亚甲基甲磺酸酯(5b)
[0123][0124]
白色固体,收率97%,分解温度115℃。
[0125]1h nmr(400mhz,cdcl3)δ7.29(d,j=8.1hz,4h),7.22(d,j=8.1hz,4h),4.09(dd,j=10.8,5.9hz,2h),3.93(dd,j=10.9,6.9hz,2h),2.82(s,6h),2.25-2.06(m,2h),1.27(s,18h).
[0126]
13
c nmr(101mhz,cdcl3)δ150.18,136.83,129.12,125.63,70.24,41.58,37.21,34.44,31.26,26.01.
[0127]
hrms(esi)calcd for[m+na,c
27h38
nao6s2]
+
:545.2002,found:545.2006.
[0128]
3,3-二(3,5-二叔丁基)苯基环丙烷-1,2-二亚甲基甲磺酸酯(5c)
[0129][0130]
白色固体,收率88%,分解温度100℃。
[0131]1h nmr(400mhz,cdcl3)δ7.23(t,j=1.8hz,2h),7.16(d,j=1.8hz,4h),4.19-4.03(m,4h),2.92(s,6h),2.15-2.07(m,2h),1.28(s,36h).
[0132]
13
c nmr(101mhz,cdcl3)δ150.96,139.47,123.56,120.88,70.39,44.04,37.61,34.81,31.41,26.80.
[0133]
hrms(maldi)calcd for[m,c
35h54
o6s2]
+
:634.3362,found:634.3355.
[0134]
3,3-二(3,5-二苯基)苯基环丙烷-1,2-二亚甲基甲磺酸酯(5d)
[0135][0136]
白色泡沫状固体,收率83%,分解温度138℃。
[0137]1h nmr(400mhz,cdcl3)δ7.69(t,j=1.7hz,2h),7.64-7.59(m,12h),7.47-7.42(m,8h),7.39-7.33(m,4h),4.28(dd,j=10.9,5.7hz,2h),4.04-3.96(m,2h),2.77(s,6h),2.43-2.32(m,2h).
[0138]
13
c nmr(101mhz,cdcl3)δ142.37,140.66,140.39,128.88,127.72,127.22,125.36,69.90,42.82,37.16,26.14.
[0139]
hrms(esi)calcd for[m+na,c
43h38
nao6s2]
+
:737.2002,found:737.2001.
[0140]
3,3-二(2-甲氧基)苯基环丙烷-1,2-二亚甲基甲磺酸酯(5e)
[0141][0142]
白色固体,收率83%,分解温度84℃。
[0143]1h nmr(400mhz,cdcl3)δ7.54(d,j=7.6hz,2h),7.18(ddd,j=8.2,7.4,1.7hz,2h),6.90(t,j=7.5hz,2h),6.73(d,j=8.2hz,2h),4.68(d,j=6.8hz,2h),3.74(s,6h),3.53(s,2h),2.96(s,6h),2.09(s,2h).
[0144]
13
c nmr(101mhz,cdcl3)δ157.99,132.25,128.55,127.04,120.19,110.33,70.92,54.98,54.91,37.69,37.55,36.00,27.21.
[0145]
hrms(esi)calcd for[m+na,c
21h30
no8s2]
+
:488.14073,found:488.14008.
[0146]
1-螺芴基环丙烷-2,3-二亚甲基甲磺酸酯(5f)
[0147][0148]
白色固体,收率81%,分解温度111℃。
[0149]1h nmr(400mhz,cdcl3)δ7.86(d,j=7.4hz,2h),7.44(td,j=7.5,1.1hz,2h),7.36(td,j=7.5,1.2hz,2h),7.23(d,j=7.7hz,2h),4.83-4.75(m,2h),4.61-4.53(m,2h),2.89(s,6h),2.63-2.55(m,2h).
[0150]
13
c nmr(101mhz,cdcl3)δ142.13,140.61,127.37,127.17,121.46,120.49,67.23,38.91,37.94,32.86.
[0151]
hrms(esi)calcd for[m+na,c
19h20
nao6s2]
+
:431.0594,found:431.0600.
[0152]
实施例4:3,3-二芳基环丙烷-1,2-二亚甲基二苯基膦1a-1f的制备
[0153]
3,3-二苯基环丙烷-1,2-二亚甲基二苯基膦1a的合成:
[0154][0155]
在氩气保护下,向100ml schlenck瓶中加入ph2ph(2.5ml,14.4mmol,2.4equiv),四氢呋喃(35ml),在-78℃及搅拌条件下,向体系中滴加正丁基锂(2.5m in hexane,7.2ml,18mmol,3equiv)。滴加完毕后,移去冷浴。室温搅拌1h后,向体系中滴加溶于四氢呋喃(15ml)的原料5a(2.47g,6mmol),室温搅拌反应。室温搅拌3h后,向体系中加入无水甲醇淬灭反应,加入硅胶制砂,通过硅胶柱层析提纯产物(洗脱剂为pe/ea=50∶1,v/v)。最终获得
白色固体2.4g,收率67%,熔点180-182℃。
[0156]1h nmr(400mhz,cdcl3)δ7.42-7.36(m,4h),7.34-7.30(m,6h),7.27-7.18(m,18h),7.16-7.10(m,2h),1.80-1.68(m,4h),1.51-1.43(m,2h).
[0157]
13
c nmr(101mhz,cdcl3)δ142.46,138.61(dd,j=118.4,13.8hz),132.76(dd,j=81.4,19.3hz),130.44,128.46(d,j=47.7hz),128.50-128.23(m),128.12,126.22,43.10(t,j=6.5hz),29.22(d,j=12.0hz),27.66-27.40(m).
[0158]
31
p nmr(162mhz,cdcl3)δ-17.44.
[0159]
hrms(esi)calcd for[m+h,c
41h37
p2]
+
:591.2365,found:591.2368.
[0160]
以下化合物的合成方法与实施例4相同
[0161]
3,3-二(4-叔丁基)苯基环丙烷-1,2-二亚甲基二苯基膦(1b)
[0162][0163]
白色固体,收率75%,熔点123-126℃。
[0164]1h nmr(400mhz,cdcl3)δ7.43-7.38(m,4h),7.35-7.31(m,6h),7.26-7.14(m,18h),1.74(qd,j=14.3,5.9hz,4h),1.46-1.39(m,2h),1.26(s,18h).
[0165]
13
c nmr(101mhz,cdcl3)δ148.70,139.69-138.08(m),139.52,132.75(dd,j=91.4,19.2hz),130.07,128.33(d,j=56.4hz),128.44-128.15(m),124.91,42.23(t,j=6.0hz),34.33,31.36,29.41(d,j=12.7hz),27.67-27.40(m).
[0166]
31
p nmr(162mhz,cdcl3)δ-17.00.
[0167]
hrms(esi)calcd for[m+h,c
49h53
p2]
+
:703.3617,found 703.3620.
[0168]
3,3-二(3,5-二叔丁基)苯基环丙烷-1,2-二亚甲基二苯基膦(1c)
[0169][0170]
白色固体,收率67%,熔点160-162℃。
[0171]1h nmr(400mhz,cdcl3)δ7.52-7.46(m,4h),7.32(d,j=1.9hz,4h),7.30-7.27(m,6h),7.23-7.14(m,12h),1.98-1.92(m,2h),1.87-1.79(m,2h),1.26(s,38h).
[0172]
13
c nmr(101mhz,cdcl3)δ149.98,142.19,139.00(dd,j=158.4,15.4hz),133.71-132.00(m),128.36(d,j=86.6hz),128.44-128.09(m),124.92-124.88(m),119.60,44.29(t,j=4.9hz),34.81,31.52,29.72(d,j=12.5hz),28.29-28.03(m).
[0173]
31
p nmr(162mhz,cdcl3)δ-17.79.
[0174]
hrms(esi)calcd for[m+h,c
57h69
p2]
+
:815.4869,found:815.4873.
[0175]
3,3-二(3,5-二苯基)苯基环丙烷-1,2-二亚甲基二苯基膦(1d)
[0176][0177]
白色固体,收率65%,熔点115-120℃。
[0178]1h nmr(400mhz,cdcl3)7.64-7.58(m,14h),7.55-7.50(m,4h),7.46-7.39(m,8h),7.36-7.31(m,10h),7.24-7.18(m,10h),2.02(ddd,j=14.1,5.2,3.0hz,2h),1.82-1.75(m,2h),1.64-1.55(m,2h).
[0179]
13
c nmr(101mhz,cdcl3)δ143.31,141.29(d,j=31.0hz),138.58(dd,j=172.7,15.1hz),132.88(dd,j=161.1,19.3hz),128.98,128.69,128.61-128.25(m),128.09,127.36,127.29,124.32,43.33(t,j=4.9hz),29.40(d,j=13.2hz),28.01-27.76(m).
[0180]
31
p nmr(162mhz,cdcl3)δ-18.29.
[0181]
hrms(esi)calcd for[m+h,c
65h53
p2]
+
:895.3617,found:895.3620.
[0182]
3,3-二(2-甲氧基)苯基环丙烷-1,2-二亚甲基二苯基膦(1e)
[0183][0184]
白色固体,收率60%,熔点124-127℃。
[0185]1h nmr(400mhz,cdcl3)δ7.46(dt,j=7.4,3.6hz,4h),7.38-7.22(m,18h),7.09(t,j=7.8hz,2h),6.82(t,j=7.4hz,2h),6.67(d,j=8.2hz,2h),3.65(s,6h),2.23(d,j=13.6hz,2h),1.44-1.23(m,4h).
[0186]
13
c nmr(101mhz,cdcl3)δ158.82,140.26-139.16(m),133.20-132.83(m),132.63-132.27(m),129.56,128.28-127.93(m),127.41,119.63,110.20,54.82,37.95-37.56(m),31.13-30.04(m),28.70-27.61(m).
[0187]
31
p nmr(162mhz,cdcl3)δ-16.06.
[0188]
hrms(esi)calcd for[m+h,c
43h41
o2p2]
+
:651.25763,found:651.25751.
[0189]
1-螺芴基环丙烷-2,3-二亚甲基二苯基膦(1f)
[0190][0191]
白色固体,收率56%,熔点147-149℃。
[0192]1h nmr(400mhz,cdcl3)δ7.75(d,j=7.5hz,2h),7.33-7.10(m,24h),6.98(d,j=7.6hz,2h),2.42-2.31(m,4h),1.85-1.76(m,2h).
[0193]
13
c nmr(101mhz,cdcl3)δ144.72,140.40,137.98(dd,j=25.8,14.4hz),132.57
(dd,j=47.8,18.9hz),128.61-127.96(m),126.07,125.63,121.38,119.87,40.66(t,j=7.3hz),35.71-35.27(m),27.09(d,j=14.1hz).
[0194]
31
p nmr(162mhz,cdcl3)δ-16.01.
[0195]
hrms(esi)calcd for[m+h,c
41h35
p2]
+
:589.22085,found:589.22114.
[0196]
实施例5:3,3-二苯基环丙烷-1,2-二亚甲基二(3,5-二叔丁基)苯基膦1g的制备
[0197][0198]
在氩气保护下,向100ml schlenck瓶中加入二芳基膦硼烷加合物(2.15g,5mmol,2equiv),四氢呋喃(10ml),在-78℃及搅拌条件下,向体系中滴加正丁基锂(2.4m in hexane,2.5ml,6mmol,2.4equiv)。滴加完毕后,移去冷浴。室温搅拌0.5h后,向体系中滴加溶于四氢呋喃(15ml)的原料5a(1.05g,2.5mmol),室温搅拌反应。室温搅拌3h后,向体系中加入无水甲醇淬灭反应,加入硅胶制砂,通过快速硅胶柱层析过滤产物。在氩气保护下,向50ml圆底烧瓶中加入上述产物,脱气吗啉(8ml),加热至110℃,搅拌反应。11h后停止加热,待反应体系冷却至室温后真空脱溶,之后以二氯甲烷溶剂粗产物,加入硅胶制砂,通过硅胶柱层析提纯产物(洗脱剂为pe/ea=50∶1,v/v)。最终获得白色固体1.22g,收率47%,熔点88-92℃。
[0199]1h nmr(400mhz,cdcl3)δ7.40-7.34(m,10h),7.29(t,j=1.9hz,2h),7.21(t,j=7.5hz,4h),7.15-7.08(m,6h),1.90(dd,j=14.2,5.2hz,2h),1.71(dd,j=14.1,7.2hz,2h),1.62-1.53(m,2h),1.26(d,j=26.5hz,72h).
[0200]
13
c nmr(101mhz,cdcl3)δ150.47-150.20(m),142.82,137.53(dd,j=141.2,13.7hz),130.72,128.01,126.98(dd,j=73.2,20.3hz),126.13,122.42(d,j=47.4hz),44.32(t,j=6.0hz),34.91(d,j=9.5hz),31.44(d,j=6.0hz),30.10(d,j=12.9hz),27.68-27.42(m).
[0201]
31
p nmr(162mhz,cdcl3)δ-17.28.
[0202]
hrms(esi)calcd for[m+h,c
73h101
p2]
+
:1039.7373,found:1039.7378.
[0203]
实施例6:3,3-二苯基环丙烷-1,2-二亚甲基二叔丁基膦1h的制备
[0204][0205]
在氩气保护下,向25ml schlenck管中加入二叔丁基膦硼烷加合物(198mg,1.2mmol,2.4equiv),四氢呋喃(4ml),在-78℃及搅拌条件下,向体系中滴加正丁基锂(2.4m in hexane,0.63ml,1.5mmol,3equiv)。滴加完毕后,移去冷浴。室温搅拌1h后,向体系中滴加溶于四氢呋喃(2ml)的原料5a(210mg,0.5mmol),室温搅拌反应。室温搅拌3h后,向体系中加入无水甲醇淬灭反应,加入硅胶制砂,通过硅胶柱层析提纯产物(洗脱剂为pe/ea=50∶1,v/v)。最终获得白色固体220mg,收率82%。
[0206]1h nmr(400mhz,cdcl3)δ7.39(d,j=7.3hz,4h),7.30-7.22(m,4h),7.20-7.11(m,2h),1.95-1.86(m,2h),1.47-1.36(m,4h),1.16(dd,j=53.4,12.5hz,36h),0.64(d,j=115.2hz,6h).
[0207]
31
p nmr(162mhz,cdcl3)δ45.05(d,j=81.2hz).
[0208]
hrms(esi)calcd for[m+nh4,c
33h62
b2np2]
+
:556.45381,found:556.45428.
[0209]
在氩气保护下,向25ml封管中加入上述二叔丁基膦配体硼烷加合物(160mg,0.3mmol),脱气吗啉(2ml),封好体系后加热至120℃反应。12h后停止加热,待反应体系冷却至室温后,先在室温条件下真空脱溶,之后在高真空状态下加热体系以便除去吗啉硼烷加合物。将处理好的产物存放于手套箱,产物为无色粘稠油状物,通过nmr鉴定为核磁纯。
[0210]1h nmr(400mhz,cdcl3)δ7.32-7.28(m,4h),7.25-7.19(m,4h),7.14-7.09(m,2h),1.71-1.63(m,2h),1.24-1.20(m,4h),1.02(dd,j=58.5,10.7hz,36h).
[0211]
13
c nmr(101mhz,cdcl3)δ143.04,130.73,128.03,125.91,31.37(d,j=7.3hz),31.17(d,j=6.3hz),30.75(dd,j=26.2,7.1hz),29.67(dd,j=39.2,13.2hz),22.18(d,j=20.6hz).
[0212]
31
p nmr(162mhz,cdcl3)δ26.23.
[0213]
实施例7:富马酸二[(r)-联萘酚单甲醚]酯6的制备
[0214][0215]
于250ml圆底烧瓶中加入(r)-联萘酚单甲醚(2.71g,9mmol,2equiv),甲苯(50ml),三乙胺(1.56ml,11.25mmol,2.5equiv),搅拌均匀。在0℃及搅拌条件下,向体系中滴加溶于甲苯(10ml)的富马酰氯(488μl,4.5mmol),加料完毕后,移去冷浴,令体系自然恢复室温,tlc监控反应。室温搅拌4h后,原料转化完毕,向体系中加入硅胶制砂,通过硅胶柱层析提纯产物(洗脱剂为pe/dcm=1∶1,v/v)。获得淡黄色固体2.27g,收率72%,熔点230-234℃。
[0216]1h nmr(400mhz,cdcl3)δ7.94(dd,j=17.0,8.8hz,6h),7.81(d,j=8.2hz,2h),7.44(ddd,j=8.1,6.5,1.4hz,2h),7.39(d,j=8.9hz,2h),7.30-7.14(m,10h),7.04(d,j=8.5hz,2h),6.17(s,2h),3.60(s,6h).
[0217]
13
c nmr(101mhz,cdcl3)δ162.68,154.73,146.16,133.53,133.47,133.24,131.84,130.13,129.11,128.79,128.13,127.82,126.66,126.57,126.19,125.69,125.04,124.93,123.67,121.20,116.90,113.17,56.35.
[0218]
实施例8:光学活性3,3-二苯基环丙烷-1,2-二亚甲基醇4a的拆分
[0219][0220]
于100ml圆底烧瓶中加入手性富马酸酯6(1.76g,2.4mmol),二苯基重氮(711mg,3.6mmol,1.5equiv),甲苯(25ml),搅拌均匀后加热至100℃反应。7h后,停止加热,待反应体系冷却至室温后,加入硅胶制砂,通过硅胶柱层析提纯产物(洗脱剂为pe/dcm=1∶1~1∶2,v/v)。分离得到其中一个非对映异构体3g 1.23g,收率58%;另外一个非对映异构体3h 749mg,收率35%。它们的表征数据如下所述:
[0221]
(1s,2s)-3,3-二苯基环丙烷-1,2-二羧酸二[(r)-联萘酚单甲醚]酯(3g)
[0222][0223]
白色固体,熔点168-178℃。
[0224]1h nmr(400mhz,cdcl3)δ8.04(d,j=9.0hz,2h),7.90-7.82(m,6h),7.48(d,j=9.1hz,2h),7.40(ddd,j=8.2,5.9,2.1hz,2h),7.33(ddd,j=8.0,6.7,1.2hz,2h),7.27-7.19(m,6h),7.14-7.05(m,4h),7.01-6.95(m,2h),6.87(t,j=7.6hz,4h),6.47-6.44(m,4h),3.75(s,6h),2.84(s,2h).
[0225]
13
c nmr(101mhz,cdcl3)δ166.41,155.04,146.42,139.09,133.69,133.43,131.57,130.08,128.97,128.78,128.27,128.07,128.02,126.98,126.74,126.43,126.19,125.43,125.40,124.35,123.69,121.14,117.53,113.51,56.61,48.12,32.02.
[0226]
(1r,2r)-3,3-二苯基环丙烷-1,2-二羧酸二[(r)-联萘酚单甲醚]酯(3h)
[0227][0228]
白色固体,熔点165-175℃。
[0229]1h nmr(400mhz,cdcl3)δ7.97(dd,j=9.1,2.2hz,2h),7.91-7.80(m,6h),7.44-7.31(m,6h),7.29-7.24(m,6h),7.12(dd,j=8.6,4.1hz,2h),7.06-6.96(m,8h),6.62-6.57(m,4h),3.58(s,6h),2.60(s,2h).
[0230]
13
c nmr(101mhz,cdcl3)δ166.29,154.86,146.11,139.22,133.53,133.39,
131.56,130.15,129.00,128.88,128.52,128.28,127.99,127.87,126.96,126.64,126.33,126.14,125.42,124.46,123.73,121.00,117.59,113.90,56.62,47.66,31.67.
[0231]
于100ml圆底烧瓶中加入手性环丙烷二酯3g(1.23g,1.45mmol),四氢呋喃(15ml),搅拌均匀后向体系中滴加四氢呋喃(5ml)的四氢铝锂(272mg,7.25mmol,5equiv)的混合物,加料完毕后,室温搅拌反应。1h后,缓慢向体系中加入结晶硫酸钠(na2so4·
xh2o)淬灭反应,抽滤,以dcm洗涤滤饼,浓缩滤液,加入硅胶制砂,通过硅胶柱层析分离回收(r)-联萘酚单甲醚及提纯目标产物。获得(1s,2s)-4a 345mg,呈白色蜡状无定形物,收率94%。经hplc测定,拆分所得手性环丙烷二醇(1s,2s)-4a的ee值为99.7%(ic-3,nhexane/iproh=90∶10,flow rate=1.0ml/min,l=210nm)。[α]
d29
=+252.2(c 1.0,chcl3)。
[0232]
(1s,2s)-3,3-二苯基环丙烷-1,2-二亚甲基醇[(1s,2s)-4a]
[0233][0234]1h nmr(400mhz,cdcl3)δ7.33-7.24(m,8h),7.20-7.15(m,2h),3.55(dd,j=11.8,5.7hz,2h),3.25(dd,j=11.3,6.7hz,2h),2.27(br,2h),1.96(p,j=5.3hz,2h).
[0235]
13
c nmr(101mhz,cdcl3)δ141.85,129.19,128.27,126.33,63.22,40.32,29.61.
[0236]
(1r,2r)-4a的合成方法与上述(1s,2s)-4a相同。所得(1r,2r)-4a为白色蜡状无定形物,收率94%。经hplc测定,手性环丙烷二醇(1r,2r)-4a的ee值为99.1%(ic-3,nhexane/iproh=90∶10,flow rate=1.0ml/min,l=210nm)。[α]
d29
=-206.6(c 1.0,chcl3)。此外,两步反应共回收(r)-联萘酚单甲醚1.21g,回收率84%,回收的(r)-联萘酚单甲醚的ee值为99.8%(hplc:ad-h,nhexane/iproh=90∶10,flow rate=1.0ml/min,l=230nm)。
[0237]
(1r,2r)-3,3-二苯基环丙烷-1,2-二亚甲基醇[(1r,2r)-4a]
[0238][0239]1h nmr(400mhz,cdcl3)δ7.28-7.20(m,8h),7.18-7.12(m,2h),3.69(dt,j=11.7,3.8hz,2h),3.47(br,2h),3.00(ddd,j=11.4,7.8,2.6hz,2h),1.92(t,j=5.3hz,2h).
[0240]
13
c nmr(101mhz,cdcl3)δ141.93,129.34,128.46,126.53,63.34,40.69,29.62.
[0241]
实施例9:(1s,2s)-3,3-二苯基环丙烷-1,2-二亚甲基甲磺酸酯(1s,2s)-5a的制备
[0242][0243]
于250ml圆底烧瓶中加入(1s,2s)-4a(2.24g,8.8mmol),二氯甲烷(60ml),三乙胺(3.7ml,26.4mmol,3equiv),搅拌均匀。在0℃及搅拌条件下,向体系中滴加甲磺酰氯(2ml,26.4mmol,3equiv),加料完毕后,移去冷浴,室温搅拌,tlc监控反应。2h后,原料转化完毕,向体系中加入硅胶制砂,通过硅胶柱层析提纯产物(洗脱剂为pe/ea=1∶1,v/v)。最后获得白色固体产物2.8g,收率78%,产物分解温度为95℃。[α]
d29
=+178.4(c 1.0,chcl3)。
[0244]1h nmr(400mhz,cdcl3)δ7.36-7.27(m,8h),7.25-7.19(m,2h),4.18-4.06(m,2h),3.97-3.81(m,2h),2.85(s,6h),2.28-2.14(m,2h).
[0245]
13
c nmr(101mhz,cdcl3)δ139.75,129.55,128.81,127.43,69.94,42.46,37.30,25.97.
[0246]
实施例10:(1s,2s)-3,3-二苯基环丙烷-1,2-二亚甲基二苯基膦(1s,2s)-1a的制备
[0247][0248]
在氩气保护下,向50ml schlenck瓶中加入ph2ph(0.53ml,3.07mmol,2.4equiv),四氢呋喃(12ml),在-78℃及搅拌条件下,向体系中滴加正丁基锂(2.4m in hexane,1.6ml,3.84mmol,3equiv)。滴加完毕后,移去冷浴。室温搅拌1h后,向体系中滴加溶于四氢呋喃(8ml)的原料(1s,2s)-5a(528mg,1.28mmol),室温搅拌反应。室温搅拌3h后,向体系中加入无水甲醇淬灭反应,加入硅胶制砂,通过硅胶柱层析提纯产物(洗脱剂为pe/ea=50∶1,v/v)。最终获得白色固体480mg,收率63%,熔点94-95℃。比旋光度[α]
d27
=+84.0(c 0.5,chcl3)。已培养出该配体的单晶,绝对构型是通过单晶结构确定的。
[0249]1h nmr(400mhz,cdcl3)δ7.42-7.36(m,4h),7.34-7.30(m,6h),7.27-7.18(m,18h),7.16-7.10(m,2h),1.80-1.67(m,4h),1.47(dt,j=6.7,5.2hz,2h).
[0250]
13
c nmr(101mhz,cdcl3)δ142.49,139.49-138.06(m),132.76(dd,j=81.4,19.3hz),130.45,128.42(d,j=48.2hz),128.50-128.21(m),128.12,126.22,43.07(d,j=13.8hz),29.26(d,j=12.2hz),27.86-27.23(m).
[0251]
31
p nmr(162mhz,cdcl3)δ-17.45.
[0252]
表1:(1s,2s)-1a的单晶测试参数
[0253][0254][0255]
实施例11:(1s,2s)-3,3-二苯基环丙烷-1,2-二亚甲基二叔丁基膦(1s,2s)-1h的制备
[0256][0257]
在氩气保护下,向100ml schlenck管中加入二叔丁基膦硼烷加合物(425mg,2.64mmol,2.2equiv),四氢呋喃(12ml),在-78℃及搅拌条件下,向体系中滴加正丁基锂(2.5m in hexane,1.2ml,3mmol,2.5equiv)。滴加完毕后,移去冷浴。室温搅拌1h后,向体系中滴加溶于四氢呋喃(8ml)的原料(1s,2s)-5a(490mg,1.2mmol),室温搅拌反应。室温搅拌2h后,向体系中加入无水甲醇淬灭反应,加入硅胶制砂,通过硅胶柱层析提纯产物(洗脱剂为pe/ea=50∶1,v/v)。最终获得白色固体260mg,收率40%,熔点232-234℃,比旋光度[α]
d27
=+154.0(c 0.5,chcl3)。已培养出该配体的单晶,绝对构型是通过单晶结构确定的。
[0258]
在氩气保护下,向25ml封管中加入上述二叔丁基膦配体硼烷加合物(300mg,0.3mmol),脱气吗啉(3ml),封好体系后加热至120℃反应。12h后停止加热,待反应体系冷却至室温后,先在室温条件下真空脱溶,之后在高真空状态下加热体系以便出去吗啉硼烷加合物。将处理好的产物存放于手套箱,产物为无色粘稠油状物,通过nmr鉴定为核磁纯。
[0259]1h nmr(400mhz,cdcl3)δ7.32-7.28(m,1h),7.25-7.19(m,1h),7.14-7.08(m,1h),1.72-1.63(m,2h),1.24-1.19(m,1h),1.02(dd,j=58.7,10.8hz,9h).
[0260]
13
c nmr(101mhz,cdcl3)δ143.06,130.73,128.03,125.91,31.27(dd,j=21.2,6.7hz),30.93,30.60,29.68(dd,j=39.1,13.2hz),22.19(d,j=20.7hz).
[0261]
31
p nmr(162mhz,cdcl3)δ26.21.
[0262]
表2:(1s,2s)-1i的单晶测试参数
[0263][0264][0265]
实施例12:环丙烷骨架双膦配体钴配合物2a-2j的制备
[0266]
3,3-二苯基环丙烷-1,2-二亚甲基二苯基膦合二氯化钴(2a):
[0267][0268]
在充满氩气的手套箱中,向100ml schlenck瓶中加入二氯化钴(261mg,2mmol),环丙烷骨架双膦配体1a(1.24g,2.1mmol,1.05equiv),四氢呋喃(30ml),室温搅拌反应。60h后脱溶,向体系中加入溶剂(nhexane/et2o=3∶1,v/v)打浆,抽滤,以正己烷洗涤滤饼,抽干,得产物。获得蓝色粉末1.36g,收率94%,熔点312-316℃。该配合物结构已经通过单晶衍射确认。
[0269]1h nmr(400mhz,cdcl3)δ16.04,15.30,9.02,8.03,7.31,1.44,-3.49,-4.55.
[0270]
ir(kbr):3053,3022,2917,2855,1639,1572,1494,1482,1434,1332,1098,1027,739,695,507cm-1
.
[0271]
表3:2a的单晶测试参数
[0272][0273]
以下化合物的合成方法与实施例12相同
[0274]
3,3-二(4-叔丁基)苯基环丙烷-1,2-二亚甲基二苯基膦合二氯化钴(2b)
[0275][0276]
蓝色固体,收率84%,熔点321-325℃。
[0277]1h nmr(400mhz,cdcl3)δ16.04,16.00,15.18,8.79,7.93,1.51,-3.44,-4.35,-4.75.
[0278]
ir(kbr):3052,2961,2902,2865,1585,1511,1435,1363,1267,1101,1026,824,740,693,508cm-1
.
[0279]
3,3-二(3,5-二叔丁基)苯基环丙烷-1,2-二亚甲基二苯基膦合二氯化钴(2c)
[0280][0281]
蓝色固体,收率67%,熔点248-250℃。
[0282]1h nmr(400mhz,cdcl3)δ15.91,15.59,9.20,7.44,1.92,-3.21,-3.49,-5.75,-6.63.
[0283]
ir(kbr):3072,3054,2962,2904,2867,1595,1478,1435,1362,1247,1027,879,738,694,509cm-1
.
[0284]
3,3-二(3,5-二苯基)苯基环丙烷-1,2-二亚甲基二苯基膦合二氯化钴(2d)
[0285][0286]
蓝色固体,收率78%,熔点300-304℃。
[0287]1h nmr(400mhz,cdcl3)δ15.82,15.38,9.16,8.34,7.87,7.82,7.66,-3.40,-4.45,-4.92.
[0288]
ir(kbr):3053,3032,2855,1593,1577,1497,1434,1334,1027,880,758,739,695,614,497cm-1
.
[0289]
3,3-二(2-甲氧基)苯基环丙烷-1,2-二亚甲基二苯基膦合二氯化钴(2e)
[0290][0291]
蓝色固体,收率78%,熔点293-297℃。
[0292]1h nmr(400mhz,cdcl3)δ16.23,15.20,9.37,8.13,7.23,6.86,5.16,3.79,1.43,-4.10,-4.66,-5.07.
[0293]
ir(kbr):3055,2922,2854,1558,1490,1461,1434,1264,1239,1120,1027,752,739,694,506cm-1
.
[0294]
3,3-二(2-甲氧基)苯基环丙烷-1,2-二亚甲基二苯基膦合二氯化钴(2f)
[0295][0296]
蓝色固体,收率86%,熔点294-298℃。
[0297]1h nmr(400mhz,cdcl3)δ15.49,15.45,11.36,8.83,8.68,7.90,5.29,1.43,-1.32,-2.99,-3.52,-4.63.
[0298]
ir(kbr):3049,2900,2852,2777,1637,1567,1480,1447,1433,1368,1329,1026,737,695,511cm-1
.
[0299]
3,3-二苯基环丙烷-1,2-二亚甲基二(3,5-二叔丁基)苯基膦合二氯化钴(2g)
[0300][0301]
蓝色固体,收率44%,熔点148-152℃。
[0302]1h nmr(400mhz,cdcl3)δ9.18,8.21,7.39,3.59,1.77,1.18,1.11,-1.44,-8.04,-9.46.
[0303]
ir(kbr):3369,3354,2961,2923,2853,1654,1631,1468,1421,1364,1249,1029,750,706,447cm-1
.
[0304]
实施例14:环丙烷骨架双膦配体钴配合物催化1-苯基-1-丙炔的硼氢化反应
[0305][0306]
在充满氩气的手套箱中,向10ml封管中加入环丙烷骨架双膦配体钴配合物(ii)(0.006mmol,3mol%),叔丁醇钠(1.9mg,0.02mmol,10mol%),甲苯(1ml)。搅拌均匀后依次向体系中加入频哪醇硼烷(30.7mg,0.24mmol,1.2equiv),1-苯基-1-丙炔(23.2mg,0.2mmol),用旋塞封好后于室温下搅拌6小时。反应结束后,将反应液通过滴管柱过滤,取少量滤液配制gc-ms样品,通过gc-ms确定产物异构体比例,其余滤液脱溶,加入二溴甲烷作为核磁内标,通过1h nmr确定反应的转化率及收率,并确认产物中异构体比例。
[0307]
表4:环丙烷骨架双膦配体钴配合物催化1-苯基-1-丙炔硼氢化的实验结果
[0308][0309]a转化率,收率由1h nmr测定。b产物比例由gc-ms和1h nmr测定。
[0310]
实施例15:不同活化试剂下钴催化1-苯基-1-丙炔硼氢化反应情况
[0311][0312]
在充满氩气的手套箱中,向10ml封管中加入环丙烷骨架双膦配体钴配合物2a(4.3mg,0.006mmol,3mol%),甲苯(1ml),搅拌溶解后向体系中加入活化试剂(0.02mmol,10mol%)。搅拌均匀后依次向体系中加入频哪醇硼烷(30.7mg,0.24mmol,1.2equiv),1-苯基-1-丙炔(23.2mg,0.2mmol),用旋塞封好后于室温下搅拌6小时。反应结束后,将反应液通过滴管柱过滤,取少量滤液配制gc-ms样品,通过gc-ms确定产物异构体比例,其余滤液脱溶,加入二溴甲烷作为核磁内标,通过1h nmr确定反应的转化率及收率,并确认产物中异构体比例。
[0313]
表5:不同活化试剂下钴催化1-苯基-1-丙炔硼氢化反应结果
[0314][0315][0316]a转化率,收率由1h nmr测定。b产物比例由gc-ms和1h nmr测定。
[0317]
实施例16:不同溶剂中钴催化1-苯基-1-丙炔硼氢化反应情况
[0318][0319]
在充满氩气的手套箱中,向10ml封管中加入环丙烷骨架双膦配体钴配合物2a(4.3mg,0.006mmol,3mol%),叔丁醇钠(1.9mg,0.02mmol,10mol%),溶剂(1ml)。搅拌均匀后依次向体系中加入频哪醇硼烷(30.7mg,0.24mmol,1.2equiv),1-苯基-1-丙炔(23.2mg,0.2mmol),用旋塞封好后于室温下搅拌6小时。反应结束后,将反应液通过滴管柱过滤,取少量滤液配制gc-ms样品,通过gc-ms确定产物异构体比例,其余滤液脱溶,加入二溴甲烷作为核磁内标,通过1h nmr确定反应的转化率及收率,并确认产物中异构体比例。
[0320]
表6:不同溶剂中钴催化1-苯基-1-丙炔硼氢化反应结果
[0321][0322]a转化率,收率由1h nmr测定。b产物比例由gc-ms和1h nmr测定。
[0323]
实施例17:环丙烷骨架双膦配体钴配合物催化内炔硼氢化反应底物范围
[0324][0325]
在充满氩气的手套箱中,向10ml封管中加入环丙烷骨架双膦配体钴配合物2a(10.8mg,0.015mmol,3mol%),叔丁醇钠(2.9mg,0.03mmol,6mol%),甲苯(1ml)。搅拌均匀后依次向体系中加入频哪醇硼烷(76.8mg,0.6mmol,1.2equiv),相应内炔底物(0.5mmol),用旋塞封好后于室温下搅拌12小时。反应结束后,将反应液通过滴管柱过滤,取少量滤液配制gc-ms样品,通过gc-ms确定产物异构体比例,其余滤液脱溶,通过硅胶柱层析提纯产物,并通过1h nmr确认产物中异构体比例。
[0326]
表7:环丙烷骨架双膦配体钴配合物催化内炔硼氢化反应底物范围
[0327][0328][0329]a分离收率;b产物比例由nmr测定。
[0330]
以上所述的仅是本发明的优选实施方式,应当指出,对于本领域的普通技术人员来说,在不脱离发明构思的前提下,还可以做出若干变形和改进,这些都属于本发明的保护范围。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1