一种蓝色电致发光材料及其合成方法和应用
1.本发明涉及一种有机发光材料及其合成方法和应用,特别涉及一种基于三(三唑)并三嗪单元的d3‑
a/d2‑
a/d
‑
a型蓝色电致发光材料及其合成方法,还涉及其作为有机电致发光二极管的主体材料和客体发光材料的应用,属于有机电致发光材料技术领域。
技术背景
2.有机发光二极管(oled)具有响应时间短、视角宽、发光色域宽且显示色彩艳丽,已经成为下一代新型显示技术。最近,国内外机构相继推出曲面屏显示器和折叠屏手机不仅让人们看到了oled商品化之后的科技感,也让科研工作者看到了它未来的无限可能。这对于推动有机发光二极管的发展具有重大意义。
3.热活性延迟荧光材料(tadf)作为最有应用前景的有机电致发光材料,除了其不含贵重金属,能够保环境和节约资源以外,还能充分利用单/三线态激子发光,达到理论上100%的内量子效率。自从tadf材料开发以来,其优异的性能一直领跑新第一代有机电致发光材料。通常,电子给体(d)和电子受体(a)单元直接共轭连接来构建tadf分子。这类分子中的homo和lumo能级分别分布在给体和受体单元上,这使homo和lumo能级的交叠显著降低,以保证单/三线态能隙(δe
st
)较小,实现tadf性能。迄今为止,与红光和绿光tadf材料相比,蓝色电致发光材料的发展还相对落后。因此,开发高效率发光的蓝色电致发光材料具有重要的研究意义。
4.三(三唑)并三嗪具有适中的吸电子性能及易化学修饰等优势,是一种较好的半导体电子受体单元。目前有文献报道,采用稠环电子受体单元—三(三唑)并三嗪以受体,在其外围引入咔唑或吖啶等给体单元,构筑系列蓝光tadf材料。具体如(202010024470.7)以及(dyes and pigments,2021,182,108589
‑
108597)。但目前报道的d3‑
π
‑
a星型蓝色有机热活性延迟荧光(tadf)材料其发光性能有待提高。
技术实现要素:
5.针对现有技术存在的缺陷,本发明的第一个目的是在现有技术的基础上进行改进,通过对现有的tadf材料的给体单元的种类、数目及给受体单元的连接位置进行优化,构筑的一系列d3‑
a、d2‑
a和d
‑
a型蓝色电致发光材料,其最大发射波长在400~490nm区间内。
6.本发明的第二个目的是在于提供一种蓝色电致发光材料的合成方法,该方法步骤简单,反应条件温和,有利于大规模生产。
7.本发明的第三个目的是在于提供一种蓝色电致发光材料的应用,将这类蓝色电致发光材料应用于有机电致发光二极管中,作为器件的主体材料及客体发光材料,有效提高器件外量子效率,最高达到20%以上。
8.为了实现上述技术目的,本发明提供了一种蓝色电致发光材料,其具有式i、式ii、式iii或式iv结构:
[0009][0010][0011]
其中,d为
[0012][0013]
r为氢或叔丁基。
[0014]
本发明的蓝色电致发光材料以三(三唑)并三嗪为受体单元,通过改变给受体单元的连接位置,构筑对位、间位及邻位连接的d3‑
a型化合物(式i和式ii)。这类化合物具有不同的给/受体单元连接位置,进而可调节分子的单线态与三线态的能级差,实现材料的性能调控;通过进一步改变给体单元的数目,构筑d2‑
a和d
‑
a型化合物(式iii和式iv)。在这类化合物中,不同的给体单元数目可有效调节分子内电荷转移性能,进而调控材料的性能
‑
结构关系。综上所述,本发明通过优化三(三唑)并三嗪受体单元周围的给体数目、给体类型以及给体和受体单元的连接位置等,得到一系列最大发射波长在400~490nm区间内蓝色电致发光材料,同时获得发光效率更高的蓝色电致发光材料,制备的发光器件外量子效率>20%。
[0015]
作为一个优选的方案,蓝色电致发光材料具有式v或式vi结构:
[0016][0017]
优选的两种蓝色电致发光材料在20wt%掺杂的发光器件中获得最大外量子效率分别为为13.70%和21.2%,最大发光亮度分别为713.4cd m
‑2和2309cd m
‑2,要远远高于现有的类似蓝色电致发光材料。
[0018]
本发明还提供了一种蓝色电致发光材料的合成方法,其包括以下步骤:步骤1):
[0019]
反应a:将5
‑
溴苯
‑
1h
‑
四唑与三聚氯氰,在碳酸钾作用下,于2
‑
丁酮溶剂中回流反应,得到三溴代化合物;
[0020]
所述三溴代化合物具有如下结构:
[0021][0022]
或者,
[0023]
反应b:将5
‑
溴苯
‑
1h
‑
四唑和5
‑
苯基四氮唑与三聚氯氰,在碳酸钾作用下,于2
‑
丁酮溶剂中回流反应,得到二溴代化合物和一溴代化合物;
[0024]
所述一溴代化合物具有如下结构:
[0025][0026]
所述二溴代化合物具有如下结构:
[0027][0028]
步骤2):
[0029]
反应c:将二溴代化合物、一溴代化合物或三溴代化合物与9
‑
咔唑基苯硼酸频哪醇酯类化合物在四(三苯基膦)钯/碳酸钾催化作用下,于乙醇和甲苯混合溶剂中回流反应,即得蓝色电致发光材料;
[0030]
所述蓝色电致发光材料具有式i、式ii、式iii或式iv结构,且式i、式ii、式iii或式iv结构中d为:
[0031][0032]
其中,r为氢或叔丁基;
[0033]
或者,
[0034]
反应d:将二溴代化合物、一溴代化合物或三溴代化合物与吖啶类衍生物、咔唑衍生物或仲胺衍生物,在三(二亚苄基丙酮)二钯、四氟硼酸三叔丁基膦和叔丁醇钠作用下,于甲苯溶剂中回流反应,即得蓝色电致发光材料;
[0035]
所述蓝色电致发光材料具有式i、式ii、式iii或式iv结构,且式i、式ii、式iii或式iv结构中d为:
[0036][0037]
其中,r为氢或叔丁基。
[0038]
作为一个优选的方案,反应a或反应b中,回流反应的温度为85~95℃时间为36~60小时。
[0039]
作为一个优选的方案,反应b中,5
‑
溴苯
‑
1h
‑
四唑和5
‑
苯基四氮唑的摩尔比为1:2~2:1。
[0040]
作为一个优选的方案,反应c中,回流反应温度为75~85℃时间为18~30h。
[0041]
作为一个优选的方案,反应d中,回流反应温度为115~125℃时间为18~30h。
[0042]
本发明还提供了一种蓝色电致发光材料的应用,其应用于电致发光器件。
[0043]
作为一个优选的方案,蓝色电致发光材料作为电致发光器件的主体材料或客体发光材料。采用器件结构为:ito/pedot:pss(40nm)/mcpcn:x wt%dopant(10nm)/dpepo(9nm)/tmpypb(45nm)/lif(0.5nm)/al(120nm)。在这个器件结构中ito基板玻璃作阳极,pedot:pss[poly(3,4
‑
ethylenedioxythiophene)
‑
poly(styrenesulfonate)]作空穴注入层,mcpcn[9
‑
(3
‑
(9h
‑
carbazol
‑9‑
yl)phenyl)
‑
9h
‑
carbazole
‑3‑
carbonitrile]为主体材料,dpepo与tmpypb分别为空穴阻挡层和电子传输层,lif/al和liq/al为阴极。
[0044]
相对现有技术,本发明的技术方案带来的有益效果在于:
[0045]
1、本发明获得了一系列最大发射波长在深蓝光(400nm)至天蓝光(490nm)之间的tadf材料。
[0046]
2、本发明通过对tadf材料的给体单元的数目、给体和受体单元的连接位置以及给体种类等进行优化,获得发光性能最佳的深蓝色电致发光材料,制备的发光器件外量子效率>20%。
附图说明
[0047]
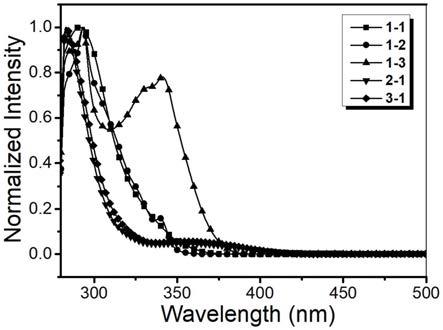
【图1】为本发明实施例1制得的化合物1
‑
1~3
‑
1在甲苯溶液中的紫外可见光吸收
光谱图。
[0048]
【图2】为本发明实施例1制得的化合物1
‑
1~3
‑
1在甲苯溶液中的光致发光光谱图。
[0049]
【图3】为本发明实施例1制得的化合物1
‑
1~3
‑
1在10%掺杂pmma薄膜中的紫外可见光吸收光谱图。
[0050]
【图4】为本发明实施例1制得的化合物在1
‑
1~3
‑
1在10%掺杂pmma薄膜中的荧光光谱。
[0051]
【图5】为本发明实施例1制得的化合物1
‑
1~1
‑
3的器件性能表征图。
[0052]
【图6】为本发明实施例1制得的化合物2
‑
1和3
‑
1的器件性能表征图。
具体实施方式
[0053]
以下具体实施案例旨在对本发明进一步说明内容,但这些具体实施方案不以任何方式限制本发明权利要求的保护范围。
[0054]
以下实施例中涉及的化学试剂如果没有特殊的说明,均为常规的市售产品。
[0055]
实施例1
[0056]
基于三唑并三嗪结构单元的tadf材料合成路线如下:
[0057][0058]
化合物1
‑
1的合成:
[0059]
将化合物ttt
‑
br(200mg,0.3mmol)、2
‑
(9
‑
咔唑基)苯硼酸频哪醇酯(665mg,1.8mmol)、四(三苯基膦)钯(51mg,0.045mmol)、碳酸钾(2mol/l,5ml)和10ml:30ml的乙醇和甲苯混合溶液加入到100ml单口瓶中,混合物在氮气保护下加热至80℃回流24h。待反应结束后,冷却至室温,减压旋蒸去除甲苯后反应液用二氯甲烷(4
×
30ml)萃取,收集的有机层依次通过水洗(50ml)、干燥、减压蒸馏除去溶剂,剩余物用二氯甲烷为洗脱剂柱层析分离得到白色固体155mg,产率45%。1h nmr(400mhz,dmso
‑
d6)δ8.13(d,j=7.7hz,6h),7.83(d,j=7.0hz,3h),7.79
‑
7.69(m,7h),7.60(dd,j=17.1,7.8hz,9h),7.31(t,j=7.7hz,6h),7.24(d,j=8.3hz,6h),7.17(t,j=7.5hz,6h),7.08(d,j=8.2hz,6h).
[0060]
化合物1
‑
2的合成:
[0061]
将化合物ttt
‑
br(180mg,0.27mmol)、3
‑
(9h
‑
咔唑
‑9‑
基)苯硼酸(256mg,0.892mmol)、四(三苯基膦)钯(46mg,0.041mmol)、碳酸钾(2mol/l,10ml)和5ml:15ml的乙醇和甲苯混合溶液加入到100ml单口瓶中,混合物在氮气保护下加热至80℃回流24h。待反应
结束后,冷却至室温,减压旋蒸去除甲苯后反应液用二氯甲烷(4
×
30ml)萃取,收集的有机层依次通过水洗(50ml)、干燥、减压蒸馏除去溶剂,剩余物用二氯甲烷为洗脱剂柱层析分离得到白色固体60mg,产率19%。1h nmr(300mhz,cdcl3)δ8.21(d,j=8.5hz,6h),8.13(d,j=7.7hz,6h),7.87(d,j=8.4hz,9h),7.74(dt,j=15.3,7.8hz,6h),7.60(d,j=7.5hz,3h),7.43(dt,j=16.1,8.0hz,12h),7.30(s,6h).
[0062]
化合物1
‑
3的合成:
[0063]
将化合物ttt
‑
br(300mg,0.45mmol)、4
‑
(9h
‑
咔唑
‑9‑
基)苯硼酸(776mg,2.7mmol)、四(三苯基膦)钯(77mg,0.068mmol)、碳酸钾(2mol/l,15ml)和10ml:30ml的乙醇和甲苯混合溶液加入到100ml单口瓶中,混合物在氮气保护下加热至80℃回流24h。待反应结束后,冷却至室温,减压旋蒸去除甲苯后反应液用二氯甲烷(4
×
30ml)萃取,收集的有机层依次通过水洗(50ml)、干燥、减压蒸馏除去溶剂,剩余物用二氯甲烷为洗脱剂柱层析分离得到白色固体100mg,产率17%。1h nmr(400mhz,dmso
‑
d6)δ8.27(dd,j=17.2,8.1hz,12h),8.17(t,j=8.6hz,12h),7.84(d,j=8.4hz,6h),7.51(q,j=8.4hz,12h),7.34(t,j=7.7hz,6h).
[0064]
化合物2和3的合成:
[0065]
在200ml单口瓶中加入5
‑
(4
‑
溴苯)
‑
1h
‑
四唑(1.2g,5.43mmol)、5
‑
苯基四氮唑(1.75g,11.95mmol)、三聚氯氰(1g,5.43mmol)无水碳酸钾(9g,65.16mmol)和100ml2
‑
丁酮,混合物在干燥空气条件下加热至90℃回流48小时。反应停止后,减压旋蒸去除2
‑
丁酮,用水洗去残留无机盐和水溶性杂质后烘干,剩余混合物加二氯甲烷溶解后加硅胶颗粒旋干,用二氯甲烷为洗脱剂经柱层析分离得到600mg化合物s
‑
ttt
‑
br和500mg化合物d
‑
ttt
‑
br,均为白色粉末。1:1h nmr(500mhz,cdcl3)δ8.14
‑
8.10(m,2h),8.05(t,j=8.8hz,4h),7.75(dd,j=8.7,2.4hz,4h),7.67(t,j=7.4hz,1h),7.61(t,j=7.4hz,2h).2:1h nmr(500mhz,cdcl3)δ8.14
‑
8.10(m,2h),8.05(t,j=8.8hz,4h),7.75(dd,j=8.7,2.4hz,4h),7.67(t,j=7.4hz,1h),7.61(t,j=7.4hz,2h).
[0066]
化合物2
‑
1的合成:
[0067]
将化合物2(120mg,0.236mmol)、9,10
‑
二氢
‑
9,9
‑
二甲基吖啶(99mg,0.472mmol)、三(二亚苄基丙酮)二钯(13mg,0.014mmol)、四氟化硼酸三叔丁基膦(8.3mg,0.028mmol)、叔丁醇钠(91mg,0.944mmol)和25ml甲苯溶液加入50ml单口瓶中,混合物溶液在氮气保护下加热至120℃回流24h。待反应结束后,冷却至室温,减压旋蒸去除甲苯,剩下的反应物固体水洗(50ml)去除水溶性杂质、干燥、减压蒸馏除去剩余溶剂,剩余物用石油醚:二氯甲烷(1:1)混合溶剂为洗脱剂柱层析分离得到白色固体60mg,产率40%。1h nmr(400mhz,cdcl3)δ8.46(d,j=8.5hz,2h),8.19
‑
8.15(m,4h),7.71
‑
7.58(m,9h),7.49(dd,j=7.6,1.4hz,2h),7.07
‑
6.95(m,5h),6.43(d,j=7.1hz,2h),1.71(s,6h).
[0068]
化合物3
‑
1的合成:
[0069]
将化合物3(120mg,0.204mmol)、9,10
‑
二氢
‑
9,9
‑
二甲基吖啶(171mg,0.817mmol)、三(二亚苄基丙酮)二钯(11.2mg,0.012mmol)、四氟化硼酸三叔丁基膦(7.1mg,0.025mmol)、叔丁醇钠(78mg,0.817mmol)和50ml甲苯溶液加入100ml单口瓶中,混合物溶液在氮气保护下加热至120℃回流24h。待反应结束后,冷却至室温,减压旋蒸去除甲苯,剩下的反应物固体水洗(50ml)去除水溶性杂质、干燥、减压蒸馏除去剩余溶剂,剩余物用石油醚:二氯甲烷(1:1)为洗脱剂柱层析分离得到黄色固体70mg,产率40%。1h nmr(400mhz,cdcl3)δ8.48(dd,
j=8.5,2.7hz,4h),8.22
‑
8.17(m,2h),7.71
‑
7.60(m,7h),7.50(d,j=7.6hz,4h),7.08
‑
7.02(m,4h),6.99(t,j=7.4hz,4h),6.47
‑
6.42(m,4h),1.71(s,12h).
[0070]
实施例2
[0071]
将化合物溶解在甲苯中配成10
‑5m溶液,测试其溶液的紫外可见吸收光谱。由图1可知,化合物1
‑
1~3
‑
1在溶液中的紫外可见吸收光谱大致有两种吸收峰:短波长(300nm)处的吸收峰主要归属于分子的π
‑
π*的跃迁吸收;长波长(330~350nm)的吸收峰归属于分子内给体单元到受体单元的电荷转移(ict)跃迁吸收峰。
[0072]
实施例3
[0073]
实施例1中的化合物1
‑
1~3
‑
1的光致发光性能测试。将化合物1
‑
1~3
‑
1溶解在甲苯中配置成10
‑5m溶液,测试其溶液的光致发光光谱,如图2所示,在光激发下,化合物从深蓝光至天蓝光区域。其中化合物1
‑
1的最大发射峰为410nm,化合物1
‑
2的发射峰为410nm,化合物1
‑
3的最大发射峰大致位于424nm,呈深蓝光发射;化合物2
‑
1的最大发射峰为468nm,化合物3
‑
1的最大发射峰为471nm,呈天蓝光发射。
[0074]
实施例4
[0075]
将实施例1的化合物1
‑
1~3
‑
1掺杂进pmma中,测试其10%wtpmma掺杂薄膜的紫外可见吸收和光致发光光谱。由图3和4可知掺杂薄膜中的吸收与溶液中大致相同,由图4可知,在10%pmma掺杂薄膜中化合物2
‑
1和3
‑
1的发射峰分别为为428nm和446nm有明显的蓝移,而化合物1
‑
1~1
‑
3发射峰没有明显的变化。
[0076]
实施例5
[0077]
实施例1中的化合物1
‑
1~3
‑
1在有机电致发光器件中的应用(具体器件结构参见说明书)。以化合物1
‑
1~3
‑
1作为器件发光层掺杂剂,通过溶液法制备电致发光器件,其中1
‑
1~3
‑
1的掺杂比例分别为1,3,5,15,20%)其中化合物1
‑
1的器件获得最大外量子效率别为3.76%,最大发光亮度分别为143.4cd m
‑2;化合物2
‑
1和化合物3
‑
1在20wt%掺杂的器件获得最大外量子效率分别为为13.70%和21.2%,最大发光亮度分别为713.4cd m
‑2和2309cd m
‑2,为如图5和6所示。
[0078]
尽管结合了优选实施例对本发明进行了说明,但本发明并不局限于上述实施例,应当理解所附权利要求概括了本发明的范围。在本发明构思的指导下,本领域的技术人员应当意识到,对本发明的各实施例方案所进行的一定的改变,都将被本发明的权利要求书的精神和范围所覆盖。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1