一种高纯度α-熊果苷及其制备方法和应用与流程
一种高纯度
α-熊果苷及其制备方法和应用
技术领域
1.本发明属于生物合成技术领域,具体涉及一种高纯度α-熊果苷及其制备方法和应用。
背景技术:
2.α-熊果苷化学名称为4-羟基苯-α-d-吡喃葡萄糖苷,其美白活性是β-熊果苷的十倍。研究发现熊果苷作为酪氨酸酶的抑制剂,能阻断多巴以及多巴醌的合成,从而有效地抑制黑色素生成,具有美白作用,且对皮肤无刺激性,毒副作用小。α-熊果苷显著抑制酪氨酸酶活性,是一种新兴的无刺激、无过敏和配伍性强的天然美白活性物质,化妆品中熊果苷推荐用量为1%~5%,具有很高的经济价值。然而,α-熊果苷对光照、温度和ph等环境因素有较强的敏感性,温度和ph过高都会导致其分解。
3.cn105671107a公开了一种利用生物酶反应和水结晶纯化制备环境友好α
‑ꢀ
熊果苷的方法,该方法存在下述缺陷:(1)该方法需要进行7次重结晶,步骤繁琐、操作周期长,进而增加生产成本,不适宜工业化生产;(2)氢醌在世界卫生组织国际癌症研究机构公布的致癌物清单的3类致癌物清单中,该方法难以有效去除氢醌,纯化后氢醌含量仍高达20ppm,同时不符合《q/jsb 001-2016 化妆品用原料α-熊果苷质量标准》规定的10ppm标准,另外,当α-熊果苷应用于质量要求更高的食品级、医药级原料时,更难以达到标准,难以保障使用的安全性。
4.现有技术中存在α-熊果苷制备工艺繁琐复杂、周期长;产品质量差、不易干燥、易分解等问题,亟需提供一种工艺简单且能够获得纯度高、晶型好、质量高的α-熊果苷的制备方法。
技术实现要素:
5.本发明的目的在于提供一种高纯度α-熊果苷的制备方法,包括下述步骤:将待精制的α-熊果苷溶解于水中,进行二次降温重结晶,分离,收集固体,干燥,即得。
6.本发明的优选技术方案中,所述二次降温重结晶在第一次降温重结晶后不进行分离,然后进行第二次降温重结晶。
7.本发明的优选技术方案中,待精制的α-熊果苷和水的重量体积比为 1:2.5-1:5;优选为1:2.8-1:4.5,更优选为1:3-1:4。
8.本发明的优选技术方案中,在进行二次降温重结晶之前,先将α-熊果苷水溶液真空浓缩至饱和状态。
9.本发明的优选技术方案中,真空浓缩时温度设置为40-85℃,优选为 45-80℃,更优选为50-75℃。
10.本发明的优选技术方案中,所述二次降温重结晶包括下述步骤:
11.(1)α-熊果苷水溶液或真空浓缩液进行首次降温,析晶;
12.(2)步骤(1)中结晶体系升温重新溶解;
13.(3)步骤(2)溶解后体系进行第二次降温,析晶。
14.本发明的优选技术方案中,析晶过程、溶解过程均在搅拌条件下进行。
15.本发明的优选技术方案中,搅拌速度为15-35r/min,优选为20-30r/min,更优选为22-28r/min。
16.本发明的优选技术方案中,步骤(1)中,降温至15-30℃,优选为20-25℃。
17.本发明的优选技术方案中,降温后维持0.5-3h,优选为1-2h。
18.本发明的优选技术方案中,步骤(2)中,升温温度为35-80℃,优选为 40-75℃,更优选为45-70℃。
19.本发明的优选技术方案中,步骤(3)中,所述第二次降温为梯度降温。
20.本发明的优选技术方案中,梯度降温至5-25℃,优选为10-15℃。
21.本发明的优选技术方案中,梯度降温的速率为每一小时降温5-10℃。
22.本发明的优选技术方案中,所述降温选自自然冷却降温、强制冷却降温的任一种或其组合。
23.本发明的优选技术方案中,所述强制冷却降温采用冷却介质对结晶体系实现强制降温。
24.本发明的优选技术方案中,所述冷却介质选自冷凝水、冰水、乙醇、乙二醇的任一种或其组合。
25.本发明的优选技术方案中,所述分离选自过滤、离心的任一种或其组合。
26.本发明的优选技术方案中,所述分离中洗涤所用溶剂为水。
27.本发明的优选技术方案中,所述干燥选自常压干燥、真空干燥或减压干燥的任一种或其组合。
28.本发明的优选技术方案中,干燥温度为25-80℃,优选为35-65℃,更优选为40-50℃。
29.本发明的优选技术方案中,所述α-熊果苷的纯度≥99.5%,优选≥99.6%,更优选≥99.7%,再优选≥99.8%,还优选≥99.9%。
30.本发明的优选技术方案中,所述α-熊果苷中氢醌含量<10ppm,优选<8ppm,更优选<6ppm,再优选<4ppm,还优选<2ppm,最优选为未检出。
31.本发明的优选技术方案中,所述α-熊果苷分离后湿料水分含量≤15%,优选≤10%,更优选≤5%。
32.本发明的优选技术方案中,所述α-熊果苷干燥后水分含量≤0.5%,优选≤0.4%,更优选≤0.3%。
33.本发明的优选技术方案中,待精制的α-熊果苷由α-熊果苷转化液经粗提取制备获得。
34.本发明的优选技术方案中,粗提取包括下述步骤:α-熊果苷转化液经过滤、分离纯化、浓缩结晶、分离,即得。
35.本发明的优选技术方案中,所述过滤选自陶瓷膜、卷式膜的任一种或其组合。
36.本发明的优选技术方案中,所述分离纯化选自色谱分离纯化。
37.本发明的优选技术方案中,所述分离选自过滤、离心的任一种或其组合。
38.本发明的优选技术方案中,所述α-熊果苷转化液的制备包括下述步骤:二糖、对苯
二酚溶于水中,在酸性条件下,经生物酶催化反应制得。
39.本发明的优选技术方案中,所述二糖选自蔗糖、麦芽糖、乳糖中的任一种或其组合。
40.本发明的优选技术方案中,所述生物酶对应于所选择的二糖,选自蔗糖酶、麦芽糖酶、乳糖酶中的任一种或其组合。
41.本发明的优选技术方案中,所述酸性条件为ph3.5-6.5,优选为ph4.0-6.0。
42.本发明的另一目的还在于提供了一种高纯度的α-熊果苷,其中,所述α-熊果苷的纯度≥99.5%,优选≥99.6%,更优选≥99.7%,再优选≥99.8%,还优选≥99.9%。
43.本发明的优选技术方案中,所述α-熊果苷中氢醌含量<10ppm,优选< 8ppm,更优选<6ppm,再优选<4ppm,还优选<2ppm,最优选为未检出。
44.本发明的优选技术方案中,所述α-熊果苷干燥后水分含量≤0.5%,优选≤0.4%,更优选≤0.3%。
45.本发明的另一目的还在于提供了一种本发明高纯度α-熊果苷在制备化妆品中的应用。
46.本发明的优选技术方案中,所述化妆品包括美白护肤品、抗氧化抗衰老护肤品。
47.本发明的另一目的还在于提供了一种本发明高纯度α-熊果苷在制备消炎制剂中的应用。
48.本发明的优选技术方案中,所述消炎制剂包括外用制剂。
49.本发明的优选技术方案中,所述外用制剂选自洗剂、软膏、乳膏、凝胶剂、酊剂、擦剂、醑剂、粉剂、油剂、糊剂、硬膏剂、涂膜剂、气雾剂、霜、乳液的任一种或其组合。
50.除非另有说明,本发明涉及液体与液体之间的百分比时,所述的百分比为体积/体积百分比;本发明涉及液体与固体之间的百分比时,所述百分比为体积/ 重量百分比;本发明涉及固体与液体之间的百分比时,所述百分比为重量/体积百分比;其余为重量/重量百分比。
51.与现有技术相比,本发明具有下述有益技术效果:
52.1、本发明的制备方法通过控制二次降温重结晶,实现了对α-熊果苷晶型的良好控制,获得了晶型好、晶体均一、大颗粒针状的α-熊果苷晶体,离心后减少了母液残留,产品纯度高、收率高;另外,该晶体容易干燥,在较低温度下即可很好地控制产品残留水分,控制在0.5%以内,不仅提高了生产效率,且很好的控制了α-熊果苷的分解,有效的控制了氢醌的含量,提高了产品质量。此外,本发明获得的α-熊果苷产品亦利于包装、运输和储藏等,减少了生产的成本。
53.2、本发明的二次降温重结晶工艺,无需添加晶种,避免了晶种的制备过程,节约生产成本;且可通过控制α-熊果苷水溶液的浓缩过饱和状态实现较快析出晶体的效果,提高了生产效率;同时在第一次析晶后重新溶解时的控制少量晶体残留,进一步实现晶体较快析出,且晶体质量更好。该过程实现了生产效率的提高、产品质量的提高。
54.3、本发明提供了一种高纯度α-熊果苷,纯度在99.9%以上,甚至高达99.99%,且本发明α-熊果苷中未检测出氢醌,含水量在0.5%以下;产品质量可控,进而保障在化妆品、医药领域应用的安全性。
附图说明
55.图1a、图1b实施例1α-熊果苷精制品外观、晶体形态图;
56.图2a、图2b对比例1α-熊果苷精制品外观、晶体形态图;
57.图3a、图3b对比例2α-熊果苷精制品外观、晶体形态图;
58.图4a、图4b对比例3α-熊果苷精制品外观、晶体形态图;
59.图5a、图5b对比例4α-熊果苷精制品外观、晶体形态图。
具体实施方式
60.下面结合实施例对本发明的技术方案作进一步详细的说明。以下实施例仅用于说明本发明而不用于限制本发明的范围。下述实施例中所使用的实验方法,如无特殊说明,均为常规方法;所用的试剂和材料,如无特殊说明,均可从商业途径获得。
61.制备例1
62.(1)α-熊果苷的制备:
63.将4t蔗糖溶解在10m3纯水中,加入400kg对苯二酚,混合均匀,调节反应溶液的ph为5,加入50kg蔗糖生物酶,搅拌至反应完全,制的α-熊果苷转化液。
64.(2)α-熊果苷的粗提取:
65.将转化液依此经过陶瓷膜、过卷式膜(800-5000分子量)过滤,得到过膜清液。过膜清液经连续色谱分离纯化、浓缩结晶,离心,得到940kg(扣除水分) α-熊果苷待精制品,纯度:89%,氢醌含量:0.5g/l。
66.实施例1
67.将94kgα-熊果苷待精制品加入300l纯水,搅拌进行溶解。60℃真空浓缩至饱和状态,25r/min搅拌,降温至25℃,维持2h,有大量细晶析出;升温至 58℃,重新溶解,大量细晶溶解,少量晶体未溶解;梯度降温至10℃(每一个小时降温8℃),搅拌5h。
68.将重结晶体系进行离心分离;在60℃,-0.095mpa真空下干燥6h;制得α
‑ꢀ
熊果苷精制品77.2kg,收率82.1%。
69.实施例2
70.将94kgα-熊果苷待精制品加入300l纯水,搅拌进行溶解。65℃真空浓缩至饱和状态,20r/min搅拌,降温至20℃,维持1h,有大量细晶析出;升温至 60℃,重新溶解,大量细晶溶解,少量晶体未溶解;梯度降温至15℃(每一个小时降温10℃),搅拌5h。
71.将重结晶体系进行离心分离;在65℃,-0.095mpa真空下干燥5h;制得α
‑ꢀ
熊果苷精制品75.3kg,收率80.1%。
72.实施例3
73.将94kgα-熊果苷待精制品加入300l纯水,搅拌进行溶解。48℃真空浓缩至饱和状态,30r/min搅拌,降温至15℃,维持2h,有大量细晶析出;升温至 43℃,重新溶解,大量细晶溶解,少量晶体未溶解;梯度降温至8℃(每一个小时降温5℃),搅拌5h。
74.将重结晶体系进行离心分离;在55℃,-0.095mpa真空下干燥8h;制得α
‑ꢀ
熊果苷精制品77.9kg,收率82.9%。
75.实施例4
76.将94kgα-熊果苷待精制品加入300l纯水,搅拌进行溶解。60℃真空浓缩至饱和状
态,50r/min搅拌,降温至20-25℃,维持3h,细晶析出;升温至25-75℃,重新溶解,大量细晶溶解,少量晶体未溶解;梯度降温至5-15℃(每一个小时降5-10℃),搅拌5h。晶体析晶速度慢,且晶体破碎。
77.对比例1
78.将9.4kgα-熊果苷待精制品加入30l纯水,搅拌进行溶解。60℃真空浓缩至饱和状态,缓慢加入8%熊果苷大颗粒针状晶种,30r/min搅拌2h,降温至10℃,析晶,搅拌5h。
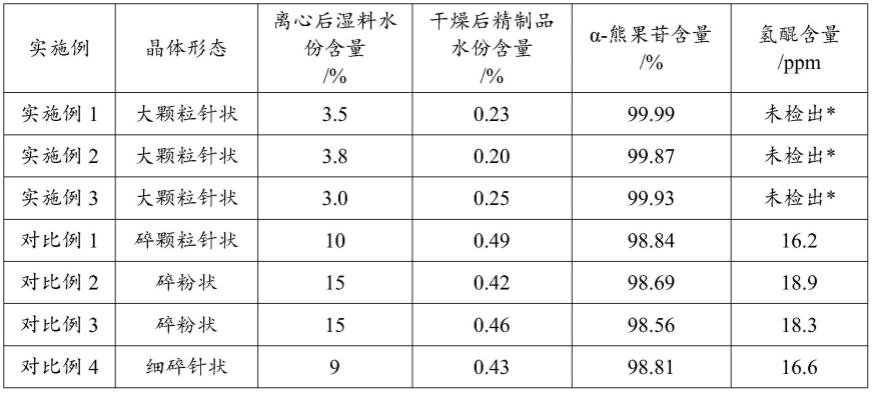
79.将重结晶体系进行离心分离;在90℃,-0.095mpa真空下干燥23h;制得α-熊果苷精制品7.14kg,收率76.0%。
80.对比例2
81.将9.4kgα-熊果苷待精制品加入30l纯水,搅拌进行溶解。60℃真空浓缩至大量晶体析出,30r/min搅拌,降温至15℃,搅拌5h。
82.将重结晶体系进行离心分离;在90℃,-0.095mpa真空下干燥26h;制得α
ꢀ‑
熊果苷精制品7.42kg,收率79%。
83.对比例3
84.将9.4kgα-熊果苷待精制品加入30l纯水,搅拌进行溶解。60℃真空浓缩至饱和状态,30r/min搅拌,缓慢加入8%熊果苷大颗粒针状晶种,继续浓缩至大量晶体析出;降温至10℃,析晶,搅拌5h。
85.将重结晶体系进行离心分离;在100℃,-0.095mpa真空下干燥20h;制得α-熊果苷精制品7.22kg,收率76.8%。
86.对比例4
87.将9.4kgα-熊果苷待精制品加入30l纯水,搅拌进行溶解。60℃真空浓缩至饱和状态,缓慢降温至10℃,析晶,搅拌5h。
88.将重结晶体系进行离心分离;在95℃,-0.095mpa真空下干燥23h;制得α
ꢀ‑
熊果苷精制品7.61kg,收率80.9%。
89.试验例1
90.性能测试:
91.对上述实施例1-3及对比例1-4制备的α-熊果苷成品进行性能测试,相关性能测试方法如下:
92.晶体形态:通过显微镜观察晶体形状;
93.水分测试:通过快速水分测定仪进行水分测定;
94.α-熊果苷含量:通过高效液相色谱法(hplc)检测α-熊果苷精制品中α
‑ꢀ
熊果苷含量;
95.氢醌含量:通过高效液相色谱法(hplc)检测α-熊果苷精制品中氢醌含量:
96.实施例1-3和对比例1-4制备的α-熊果苷精制品的检测结果如表1所示;
97.表1
[0098][0099]
未检出*:高效液相色谱法检测,氢醌含量<1ppm,甚至为0。
[0100]
以上仅是本发明的优选实施方式,本发明的保护范围并不仅局限于上述实施例,凡属于本发明思路下的技术方案均属于本发明的保护范围。应当指出,对于本技术领域的普通技术人员来说,在不脱离本发明原理前提下的若干改进和润饰,应视为本发明的保护范围。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1