一种早发性白发遗传风险基因检测试剂盒、以及一种早发性白发遗传风险评估系统和方法与流程
1.本发明属于早发性白发遗传风险评估领域,更具体地涉及一种早发性白发遗传风险基因检测试剂盒、以及一种早发性白发遗传风险评估系统和方法。
背景技术:
2.早发性白发(phg,premature graying of hair),亦即少白头,是一种高度遗传性疾病,多数定义为个体20岁前出现白发的现象,且根据白发占总发量的比例来界定其严重程度。根据研究数据表明,中国人群中约有5%~30%有过不同程度的早发性白发,且在近代人群中比例呈上升趋势。
3.由于早发性白发发病年龄的特殊性,其对青少年发育过程中的日常生活、心理健康及社交可能造成诸多负面影响。早发性白发尚无明确的治疗方案,因此导致多数早发性白发患者于发育期持续使用染发剂,从而可能对其发育甚至后期健康造成不良后果。且除青少年自身影响之外,早发性白发也会对家庭造成一定程度的心理及经济负担。
4.除遗传因素外,早发性白发的发病也受到诸多环境因素调控,例如吸烟、肥胖、氧化应激、压力、心理障碍(情绪应急、焦虑、抑郁)、酒精摄入等。鉴于早发性白发发病后对青少年及其家庭影响重大,且发病后无有效方案、难治愈的特性,急需一个能够及早发现青少年个体早发性白发风险的方法,从而进行提前的有效干预降低发病率。
5.全基因组关联研究(gwas)可以确定单核苷酸多态性(single nucleotide polymorphism,snp)与表型性状(例如本发明中的早发性白发)之间的关联。gwas方法普遍应用于疾病健康领域,已经识别了很多常见的复杂疾病相关联的遗传变异。大部分遗传变异对于疾病风险贡献通常很小,不足以单独直接预测疾病状态,但是它们的累积效应却对于疾病的风险有着更强区分和评估效果。
6.多基因风险打分(polygenic risk score,prs)是一种统计学的方法,可以根据个体的基因型谱来评估某一疾病或者性状的遗传风险,也就是多个风险位点的累积效应。利用大量的snp来计算prs对于疾病状态的预测能力更佳,但大量基因位点的检测使其检测成本高昂,不适合大规模风险筛查,因此如何找到复杂疾病风险的最佳预测prs,已经成为目前研究的重点。选择更合适的候选基因位点和prs构建方法来整合这些遗传变异构建更高预测性的早发性白发多基因遗传风险评分将有助于开展早发性白发早期风险预测。
7.目前尚未有机构采用基因检测方法对早发性白发易感性进行检测。基因分型的常见方法有:taqman探针法、snapshot法、飞行时间质谱(maldi
‑
tof ms)分型、hrm(高分辨率熔解曲线)分型等技术平台。其中,基于飞行时间质谱(maldi
‑
tof)完成的snp检测准确率可达99.9%,除了准确性高、灵活性强、通量大、检测周期短等优势外,最有吸引力的应该还是它的性价比。飞行时间质谱平台(maldi
‑
tof)是国际通用的基因单核苷酸多态性(snp)的研究平台,该方法凭借其科学性和准确性已经成为该领域的新标准。下表1给出了一些snp分型方法的优缺点。
8.表1
技术实现要素:
9.本发明目的在于提供一种早发性白发遗传风险基因检测试剂盒、以及一种早发性白发遗传风险评估系统和方法,从而解决现有技术中缺乏早发性白发早期风险预测方法的问题。
10.为了解决上述问题,本发明采用以下技术方案:
11.根据本发明的第一方面,提供一种对早发性白发易感位点进行基因分型的试剂盒,以及在评估是否具有患早发性白发的遗传风险和评估早发性白发严重程度方面的应用。本试剂盒可同时对早发性白发易感风险相关的16个基因位点进行snp分型,包括用于扩增16个基因位点的16对pcr扩增引物对。16个基因位点如下所示:rs12563397,rs16886165,rs10461617,rs3127159,rs7775323,rs7803075,rs16888276,rs1881129,rs644383,rs676856,rs17335506,rs57406257,rs9651934,rs2058343,rs117283943,rs150271955。
12.序列具体如下所示,扩增和延伸引物序列方向均为5’端至3’端,涉及的snp位置参考hg19,rs编号来自db151。其中,16个基因位点以及16对pcr扩增引物序列具体如下所示:1扩增引物对1号(1:214756124位点):g
‑‑‑
>a(表示野生型为g,突变型为a,下同)seq id no.1(上游引物,下同):cccatccctgttttaactagtseq id no.2(下游引物,下同):gctaataataataatcttttg
2扩增引物对2号(5:56023083位点):t
‑‑‑
>g(表示野生型为t,突变型为g,下同)seq id no.3(上游引物,下同):atatagttggtgctcagtaaaseq id no.4(下游引物,下同):cattttggttacttgaggtca3扩增引物对3号(5:56104308位点):g
‑‑‑
>a(表示野生型为g,突变型为a,下同)seq id no.5(上游引物,下同):gggccttcaaatgtcaaaaagseq id no.6(下游引物,下同):atccgctaacttgtcaaatat4扩增引物对4号(6:159261934位点):c
‑‑‑
>g(表示野生型为c,突变型为g,下同)seq id no.7(上游引物,下同):aagtacagtttagtgggcaggseq id no.8(下游引物,下同):cagaggagttgctggtctggc5扩增引物对5号(6:22722969位点):c
‑‑‑
>t(表示野生型为c,突变型为t,下同)seq id no.9(上游引物,下同):tgagaccctgtctcaaaaataseq id no.10(下游引物,下同):tgtggcatttggggtaaggtc6扩增引物对6号(7:130742066位点):a
‑‑‑
>g(表示野生型为a,突变型为g,下同)seq id no.11(上游引物,下同):tttgatctccttagactctctseq id no.12(下游引物,下同):tttctgctcaagcttagttcc7扩增引物对7号(8:117306606位点):t
‑‑‑
>g(表示野生型为t,突变型为g,下同)seq id no.13(上游引物,下同):cctttctccgacctcaaacatseq id no.14(下游引物,下同):aatgaattagagctacctgca8扩增引物对8号(8:117334573位点):a
‑‑‑
>g(表示野生型为a,突变型为g,下同)seq id no.15(上游引物,下同):acctcaagctcaagctggttgseq id no.16(下游引物,下同):tgcaaagtcacccttcctttt9扩增引物对9号(9:208665位点):g
‑‑‑
>a(表示野生型为g,突变型为a,下同)seq id no.17(上游引物,下同):tgggaagaatatgattttttcseq id no.18(下游引物,下同):aagcatagaacagagcccaac10扩增引物对10号(11:69329784位点):c
‑‑‑
>a(表示野生型为c,突变型为a,下同)seq id no.19(上游引物,下同):acttgcctaggaacacacagcseq id no.20(下游引物,下同):cacatggagacagggaaacgt11扩增引物对11号(12:88519346位点):t
‑‑‑
>c(表示野生型为t,突变型为c,下同)seq id no.21(上游引物,下同):ttaccattttgtacacttcaaseq id no.22(下游引物,下同):tgtttatggcttctatacttc12扩增引物对12号(12:89239190位点):g
‑‑‑
>a(表示野生型为g,突变型为a,下同)seq id no.23(上游引物,下同):gcaagctttggattagagcaaseq id no.24(下游引物,下同):taatgtgtaaattcatttctt13扩增引物对13号(12:89355709位点):c
‑‑‑
>a(表示野生型为c,突变型为a,下同)seq id no.25(上游引物,下同):tcctaatgtaccacaaaagca
seq id no.26(下游引物,下同):ctctggagtctctatttgcac14扩增引物对14号(12:89366763位点):g
‑‑‑
>a(表示野生型为g,突变型为a,下同)seq id no.27(上游引物,下同):gtgggctcacaagcaatttatseq id no.28(下游引物,下同):ttgcacataaggaagacttaa15扩增引物对15号(22:31150167位点):c
‑‑‑
>t(表示野生型为c,突变型为t,下同)seq id no.29(上游引物,下同):cttggggaaaaaatagattttseq id no.30(下游引物,下同):ctccctctgtttatagttcct16扩增引物对16号(22:31458617位点):g
‑‑‑
>a(表示野生型为g,突变型为a,下同)seq id no.31(上游引物,下同):agggtattttgcaaataagtaseq id no.32(下游引物,下同):gacacaggagaccagaacagc。
13.根据本发明的一个优选方案,该试剂盒还包括用于鉴定16个基因位点突变的16条延伸引物。16条延伸引物序列具体如下所示:seq id no.33:ccctgcctccagagtcgagtcctacgseq id no.34:aaagaaacttgcccaaggccagctttseq id no.35:tgatgttggaaacaacaccactttgaseq id no.36:ttggctggggaatgcaatcacaggacseq id no.37:tctcattctcagctattccaattgtcseq id no.38:tgaactgcgctcctggatcttttacaseq id no.39:cggtaagtatatgttttctttgatttseq id no.40:aattcagagaaacttctgggctcctaseq id no.41:gaaggaaattaatttttttccatagaseq id no.42:gacactgagtctcacaaccccacagcseq id no.43:ctgctgttactccaatactgaaagatseq id no.44:agcaatataagctgaggtggatatagseq id no.45:tctagtgagattaaaactgaatgcacseq id no.46:tcatctgtaagatgggaatcctaaaaseq id no.47:cagccaaaaattgggcagacttttccseq id no.48:ctccttgaaaaatcgtaaaagaactg
14.根据本发明,优选地,试剂盒的pcr扩增的反应体系如下:
15.根据本发明,优选地,试剂盒还包括sap反应体系,所述sap反应体系如下:sap缓冲液
ꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀ
0.17μlsap酶
ꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀ
0.5uddh2o
ꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀ
补齐至2μl。
16.所述试剂盒采用飞行时间质谱仪进行检测。首先通过多重pcr在一个体系内同时扩增目标序列,然后加入snp序列特异延伸引物,在snp位点上,延伸1个碱基。将制备的样品分析物与芯片基质共结晶后在质谱仪的真空管经强激光激发,核酸分子解吸附为单电荷离子,电场中离子飞行时间与离子质量成反比,通过检测核酸分子在真空管中的飞行时间而获得样品分析物的精确分子量,从而检测出snp位点信息。
17.根据本发明提供的试剂盒,所涉及的基因位点是基于大规模中国汉族人群早发性白发队列研究产生的基因分子位点,不涉及任何已有文献和研究;根据收集的相关数据选择出最有早发性白发预测性的一组snp作为基因分子标记。挑选位点的具体方法出自早发性白发遗传风险预测模型的构建方法,其选择出的最优模型的特征集合中包含的snp即作为试剂盒的待测分子;试剂盒涉及的snp位点集合均为最小等位基因频率不小于千分之一的普通变异位点,保证了试剂盒广泛的人群适用性;设计对应的高特异性的引物组,能对早发性白发相关风险基因进行精准基因分型。使用飞行时间质谱仪的方法确保了检测结果的准确性和灵敏度,简单可行。
18.根据本发明的第二方面,提供一种早发性白发遗传风险评估系统,包括:获取模块,用于获取测试者的dna样本;检测模块,包括如上面所述的早发性白发遗传风险基因检测试剂盒,用于对所述dna样本进行基因分型;预测模块,包括一种早发性白发遗传风险预测模型,用于对测试者的早发性白发遗传风险进行评分;以及评估模块,包括一种早发性白发遗传风险等级划分体系,用于确定测试者的早发性白发遗传风险等级。
19.根据本发明的一个优选方案,所述早发性白发遗传风险预测模型利用全基因组关联分析和机器学习算法,整合风险基因位点信息来对早发性白发遗传风险进行预测和量化打分。
20.该早发性白发遗传风险预测模型的主要使用场景是对早发性白发遗传风险基因检测试剂盒测得的基因分型数据,结合性别信息对早发性白发遗传风险进行风险评分。在被检测者全基因组数据可用和性别可知,且包含模型特征中涉及到的基因位点等其他场景时,也可使用该模型进行风险评分。
21.该早发性白发遗传风险预测模型在样本量充足的独立群体中进行了性能评估,其auc值为0.68,在目前多基因预测领域中,当前auc值为优秀水平;根据100分位数分组方式,
最高百分位数和中间分位数之间的case/control的比值比(or)为11.66,相对风险比值(rr)为2.37;在选择分类阈值为0.5时,敏感度(sensitivity)为0.89,阳性正确率(ppv)为0.67;整体准确性(accuracy):为0.66。提高分类阈值线,可提高早发性白发群体的检测敏感性和阳性正确率。
22.本发明的早发性白发遗传风险预测模型对早发性白发的严重程度的人群也具有预测区分能力(对“稍微有些白发”人群,其auc为0.65;对“接近一般白发”人群,其auc为0.69;对“大部分头发变白”人群,其auc为0.69)。基于双变量正态分布估计每个多基因评分分位数内的早发性白发比例与实际分布基本一致,可提供较为客观的早发性白发绝对风险。
23.本发明的早发性白发遗传风险预测模型所包含的特征id和模型权重和风险等位基因信息如下表2所示(涉及的snp位置参考hg19,rs编号来自db151)。
24.表2表2
25.根据本发明提供的早发性白发遗传风险预测模型,使用性别和基因位点在内的共计17个特征和一个权重为1的截距项
‑
1.25867205,性别权重值为
‑
0.60813851,所有特征进行“风险等位剂量加权求和”来作为早发性白发遗传风险分数。在使用模型时,严格根据风险等位基因在基因型重的剂量个数进行编码(例如rs12563397,风险等位基因为a,基因型ga编码为1;基因型为aa编码为2;基因型为gg编码为0。
26.根据本发明提供的这样一种早发性白发遗传风险预测模型,所使用的特征和权重是基于大数据和机器学习及基因组学相关方法训练得到,以数据为驱动,以高预测性为目
标,避免了直接使用已有研究的一些特征和权重带来的偏差;多基因分数结合性别、年龄或其他环境因素是当前风险评分的主要研究方向;在独立人群队列中进行了严格性能评估,验证模型在风险评估中的区分能力和适用范围。
27.根据本发明的一个优选方案,所采用的早发性白发遗传风险等级划分体系通过对早发性白发遗传风险预测模型得到的风险分数进行切割分组,且根据所收集到的数据集合的早发性白发流行率提示其风险程度。
28.分割区间为:等级1:[0
‑
0.201297176117716):低风险;等级2:[0.201297176117716
‑
0.281098927835626):较低风险;等级3:[0.281098927835626
‑
0.363050852087241):一般风险;等级4:[0.363050852087241
‑
0.453887538258589):较高风险;等级5:[0.453887538258589
‑
0.566654292179563):高风险;等级6:[0.566654292179563
‑
1):极高风险。风险等级从低到高共6个级别。
[0029]
该体系提供一般人群中的整体早发性白发风险等级占比分布和特定风险等级下的人群实际已患早发性白发的比例。具体如下:等级1:10.9%;等级2:18.3%;等级3:23.8%;等级4:22.7%;等级5:17%;等级6:7.3%。
[0030]
各个特定风险等级中的人群的早发性白发情况实际分布如下表3所示。
[0031]
表3
[0032]
根据本发明提供的这样一种早发性白发遗传风险等级划分体系,对模型得到的早发性白发遗传风险分数划分个体风险等级和程度说明,提供一个宏观视角下的中国人群中的整体风险分布和相同风险等级下的人群实际已患早发性白发的比例。该体系不仅提供了一个定性的风险程度预警结果,也可以提供更多基于实际数据得到的数字式定量分布结果,让被检测者更好感知自身风险。
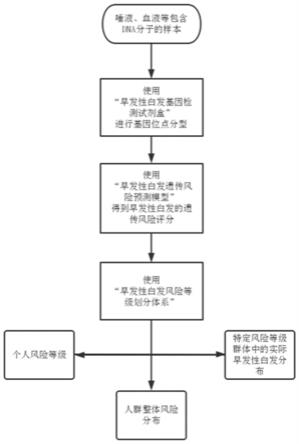
[0033]
根据本发明的第三方面,提供一种早发性白发遗传风险评估方法,如图1所示,包括以下步骤:s1:获取测试者的dna样本;s2:使用如上面所述的早发性白发基因检测试剂盒对所述dna样本进行基因分型;s3:构建一种早发性白发遗传风险预测模型,使用所述早发性白发遗传风险预测模型获得测试者的早发性白发遗传风险评分;s4:使用早发性白发风险等级划分体系,根据测试者的不同,确定个人风险等级,或人群整体风险分布,或特定风险等级群体中的实际早发性白发分布。
[0034]
步骤s3中,所述早发性白发遗传风险预测模型的构建包括以下步骤:s31:通过早发性白发调查问卷方式获取志愿者的早发性白发表型数据;s32:对参加早发性白发问卷调查的志愿者获取基因数据;s33:对收集到的问卷和基因数据进行预处理和集合划分,依据
机器学习处理模式,划分训练集合、验证集合和测试集合;s34:对训练集合进行全基因组关联分析,得到早发性白发gwas统计数据,gwas统计数据包括位点编号及其与早发性白发关联的显著性p值;s35:根据gwas统计数据,基于位点连锁相关性r2和显著性p值进行两两组合,进行位点集合过滤;s36:基于每个过滤后的位点集合,使用机器学习中的特征选择算法自适应的进行特征选择,使用经特征选择算法过滤后的特征,在训练集合中运用逻辑回归算法,建立早发性白发遗传风险预测模型,并在独立的验证集合上进行预测和性能评估;s37:基于在验证集合上的预测表现,根据auc值选择在验证集合上表现最佳的模型和最优的基因位点;s38:将最佳模型运用到独立的测试集合中进行预测,得到其模型在一般人群中的预测性能、风险划分能力和人群风险分布。
[0035]
优选地,步骤s32包括:使用genetitan多通道仪器平台在基于axiom精密医学研究阵列的高通量基因芯片上进行基因分型,实现所述基因数据的获取。
[0036]
根据本发明的第四方面,还提供一种早发性白发遗传风险评估系统在评估早发性白发遗传风险水平中的应用。
[0037]
根据本发明,首先提供了一种早发性白发基因检测试剂盒,克服了现有技术缺乏早发性白发基因检测方法的问题,具有检测效能高、检测通量大、检测成本低的优点,以实现早发性白发遗传风险预测;其次,通过大规模的早发性白发队列人群的全基因组关联性分析(gwas),使用机器学习方法(ml)构建和选择最优的遗传风险模型,提供了一种专门针对中国人群的早发性白发遗传风险水平评估方法;为了让模型更好的发挥实用价值,本发明提供了与模型配套使用的试剂盒基因检测方案,克服了遗传风险评估与基因分型间的不匹配、不兼容等不足,具有检测通量大、检测成本低的特点,以解决大规模人群的早发性白发遗传风险水平普筛,做到早发性白发的早发现、早预防和早治疗。从基因水平深度挖掘其早发性白发相关遗传信息,来区分早发性白发高、中和低风险人群。
[0038]
综上所述,根据本发明提供的一种早发性白发基因检测试剂盒、一种早发性白发遗传风险评估系统、以及一种早发性白发遗传风险评估方法,相比于现有技术具有以下显著的优越性:
[0039]
1)截止到本发明,现有技术都尚未出现对早发性白发进行遗传风险预测、划分的模型和对早发性白发进行基因检测的试剂盒。本发明根据大规模的早发性白发(大于2.5万人参与,迄今为止,最大规模的中国人群队列早发性白发研究)中国队列人群的遗传研究结果,发现的易感位点和模型使用的位点集合更适合中国人群;所有均为数据驱动,不涉及和基于公共数据集和别人已发表研究。我们目前提出的新的位点组合在任何一篇文献中都未提到过,是通过大量原始数据分析获得;
[0040]
2)使用机器学习算法,在训练和验证两个独立集合实施特征选择和最优模型选择算法构建风险预测模型,相比传统的prs模型构建方法,具有更高的预测性能和更少的模型位点。在大样本量的队列集合中展示模型总体风险预测性能和风险分布。配套早发性白发遗传风险划分体系和大样本量的队列集合,提供了客观的早发性白发绝对风险尺度和相对风险等不同维度的风险。在该集合中在最低分位数组的早发性白发仅占3%;最高分位数组中早发性白发占据高达83%;整体趋势为处于分位数越高人群中的早发性白发占比越高。根据大样本量的队列集合给出的绝对风险与基于双变量正态分布估计每个多基因评分分位数内的绝对风险基本一致。该模型不仅可以对早发性白发人群具有很好的预测性,对早
发性白发的严重程度也具有较高的预测能力;
[0041]
3)现有研究对早发性白发的遗传机制和白发背后的衰老、代谢、免疫相关等机制均不清楚,本发明涉及的snp位点和位点对应的连锁区域、基因、基因通路将会对早发性白发的遗传相关和其背后的高度相关疾病的研究提供科研价值;
[0042]
4)本发明采用的飞行时间质谱检测法,相较于荧光定量pcr、基因芯片、sanger测序、ngs等技术,具有快速、灵敏度高、成本低、简单易行的特点,适用于多基因、多位点的检测。仅使用唾液样本测试来预测早发性白发遗传风险情况。snp分型准确率高,检出率可达95%以上,准确率98%以上。附图说明(请将图5~图9中的英文修改为中文,以符合专利申请文件附图的要求)
[0043]
图1示出了根据本发明提供的一种早发性白发遗传风险评估方法的逻辑图;
[0044]
图2示出了早发性白发遗传风险预测模型的构建流程及基因检测试剂盒设计流程的详细技术示意图,其中特别明确了早发性白发易感基因位点集来源和方法;
[0045]
图3示出了早发性白发研究调查问卷逻辑图;
[0046]
图4示出了gwas质量控制流程图;
[0047]
图5中的a和b分别示出了gwas统计数据的曼哈顿图、q
‑
q图;
[0048]
图6示出了在由早发性白发和非早发性白发人群组成的独立测试集合中,case(早发性白发,右边灰色)和control(非早发性白发,左边黑色)群体的遗传风险得分分布的箱线图,纵轴为基因风险分数,图中两个分组中的小圆点代表了该组基因风险分数的平均值;
[0049]
图7示出了在由早发性白发和非早发性白发样本,年龄在20
‑
60岁组成的集合中,模型的预测分数的50分位数和早发性白发占比在不同性别中的分布图;
[0050]
图8示出了在由早发性白发和非早发性白发样本,年龄在20
‑
60岁组成的集合中,模型的预测分数的50分位数和早发性白发占比的关系图;
[0051]
图9示出了由早发性白发和非早发性白发样本,年龄在20
‑
60岁组成的集合给出的50分数下的实际绝对风险与基于双变量正态分布估计的50分位数内的绝对风险关系图;
[0052]
图10示出了本发明的试剂盒的引物设计示意图。
具体实施方式
[0053]
为了使本发明的目的、技术方案及优点更加清楚,以下结合实施例,对本发明进行进一步详细说明。应当理解,此处所描述的具体实施例仅仅用以解释本发明,并不用于限定本发明。
[0054]
实施例1:构建一种早发性白发遗传风险预测模型
[0055]
该实施例提供了一种用于评估早发性白发遗传风险的预测模型,其涵盖中国汉族人群的早发性白发基因多态性位点,包括人基因组dna的16个snp位点和性别特征。本发明的早发性白发遗传风险评估模型构建步骤如图2所示。
[0056]
1)获取早发性白发表型数据。设计调查问卷,进行推送和发放问卷,研究志愿者自愿填写问卷,进行早发性白发表型数据的收集。
[0057]
2)对参加问卷调查的参与者获取基因数据。通过snp芯片或测序经生物信息分析手段获得全基因组范围内的位点及其基因型信息。
[0058]
3)对收集到的问卷和基因数据进行预处理和集合划分。根据问卷填写数据,根据
问卷的设计逻辑进行质控后,采用“案例对照组”的实验范式,将从未有过早发性白发作为对照组,有过早发性白发作为案例组。依据机器学习处理模式,划分训练集合、验证集合和测试集合。三个集合互相独立,不包含相同的个体。
[0059]
4)对训练集合进行全基因组关联分析,得到早发性白发gwas统计数据,gwas统计数据包括位点编号及其与早发性白发关联的显著性p值。
[0060]
5)根据gwas统计数据,基于位点连锁相关性r2和显著性p值进行两两组合(p_t策略)进行位点集合过滤。
[0061]
6)基于每个过滤后的位点集合,使用机器学习中的特征选择算法自适应的进行特征选择(包含snp和gender)。使用经特征选择算法过滤后的特征,在训练集合中运用逻辑回归算法,建立早发性白发的风险预测模型,并在独立的验证集合上进行预测和性能评估。
[0062]
7)基于在验证集合上的预测表现,根据auc值选择在验证集合上表现最佳的模型和最优的基因位点。
[0063]
8)将最佳模型运用到独立的测试集合中进行预测,得到其模型在一般人群中的预测性能、风险划分能力和人群风险分布。
[0064]
在步骤1)中,设计早发性白发调查问卷,问题逻辑图为图3所示:其中。问卷形式可以是线下问卷(滚轴填写形式改成手填即可),也可以是网上电子问卷形式。此实施例采用网络电子问卷,可选择性地搭载在app中,命名为“早发性白发问卷”进行受试者的招募。受试者需完成问卷的填写。共收集到24431份的问卷。其中男女分别为11561/12870,年龄范围为2岁到121岁。数据可导出成txt文件。并不仅限于图3中所有问题,但优选包括上述问题。
[0065]
在步骤2)中,参与早发性白发调查问卷的参与者将2ml的唾液存放到唾液保存管中后,将会送到实验室,纯化唾液中的dna,并使用genetitan多通道仪器平台(thermo fisher scientific)在基于axiom精密医学研究阵列(affymetrix)的高通量基因芯片上进行基因分型。该基因芯片涵盖22个常染色体,2条性染色体和线粒体基因组的近90万个snp。通过affy基因数据生产pipeline处理后,数据以plink的二进制格式进行输出。
[0066]
在步骤3)中,分别进行问卷数据和基因数据的预处理和质量控制。对于问卷数据,使用开源统计软件r任何一个版本对问卷结果进行数据的质控和处理。
[0067]
步骤3)
‑
1:抽取问卷中的“年纪”“你是否有过早发性白发?”,“你什么时候开始长了白发?”这几个问题数据。过滤12
‑
70的样本。按照以下条件排除不可靠回答并进行分类:
[0068]
排除30岁之后早发性白发,回答:在问题“你是否有过早发性白发”为肯定回答的,但“你什么时候开始长了白发”选择了“30岁之后”的样本。
[0069]
大于30岁的人不清楚自己什么时候开始早发性白发的,认为不是早发性白发:在问题“你是否有过早发性白发”为肯定回答的,但“你什么时候开始长了白发”选择了“记不清楚了”且“年纪”大于等于30岁的人,认为不是早发性白发。
[0070]
排除不清楚自身早发性白发情况的样本:在问题“你是否有过早发性白发”中回答了“不清楚”的样本。
[0071]
在问题“你是否有过早发性白发”为肯定回答的,作为早发性白发群体即案例组(case),编码成1;在问题“你是否有过早发性白发”为否定回答的作为非早发性白发群体即对照组(control),编码成0。
[0072]
区分自然白头和早发性白发的年龄,虽在亚洲群体一般选取20岁前出现白发作为
早发性白发时间区分点,但本发明基于1)我们数据显示20岁和30岁作为时间区分点,其遗传机制基本一致;2)选择30岁可一定程度扩大case数量,增大gwas的检测效能。所以本发明设定为不大于30岁认定为早发性白发,而非自然白头。
[0073]
处理完成后共计22477份填写,其中早发性白发占比为32%。
[0074]
步骤3)
‑
2:将处理好的早发性白发表型数据进行数据集合的划分,按照7&1&2划分成表型数据的训练集合、验证集合和测试集合。划分数据集合时,使用分层抽样的方法,可以使用r的第三方扩展包“caret”的函数createfolds(),依据包含是否早发性白发信息的字段进行分层,使得三个数据集合的case vs control比例一致,不存在分布偏差。
[0075]
步骤3)
‑
3:根据处理后的问卷数据的样本编号分别匹配基因数据,可使用生物信息常用软件plink
‑‑
keep命令基于样本信息进行基因数据的过滤。划分完成后训练集合为15733人,测试集合为4496人,验证集合为2248人。基因训练集合的质量控制具体参考图4所示,其所有与常染色体相关的操作均可在plink软件中选择对应命令完成,所有与非伪常染位点相关操作通过“xwas”软件对应的命令完成。简单的包括去除maf小于0.001的位点,去除样本缺失率和位点缺失率大于0.02的样本和位点,去除不满足哈温平衡的位点,去除亲缘关系密切的样本,去除非中国汉族的样本,使用mds分析,而非pca分析。质量控制后,常染色体和伪常染snp总数为394624。非伪常染snp个数为6945。均未进行impute,均仅使用原始分型数据进行后续分析。
[0076]
在步骤4)中,将表型训练集合和基因训练集合进行全基因组关联分析,采用病例对照编码形式,使用逻辑回归(logistic)算法和基因加和(addtive)模型,将含有2个正常等位基因的snp视为该snp的风险等位基因的数量为0;将含有1个正常等位基因的snp视为该snp的风险等位基因的数量为1;将含有0个正常等位基因的snp视为该snp的风险等位基因的数量为2。协变量为人群主成分pc1
‑
pc5和性别来控制人群分层和性别分层导致的假阳性关联。得到的早发性白发gwas统计数据,其中包含早发性白发关联的显著性p值。将作为下一步位点过滤的关键指标。仅使用常染色体和伪常染snp进行后续分析。gwas统计数据的曼哈顿图、q
‑
q图如图5所示。
[0077]
在步骤5)中,使用pruning and threading方法基于上述早发性白发gwas统计数据的p值及训练集合位点间的连锁r2来筛选位点集合,筛选标准为p值可设置5e
‑
4、5e
‑
5、5e
‑
8,连锁r 2值可设置0.2、0.5、0.8、0.95,两个筛选标准两两组合共计12种(3*4)来筛选snp集合。每种组合都会得到一组基因位点集合。其结果如下表4所示。
[0078]
表4位点个数筛选标准组合2645*10^
‑
4_0.22915*10^
‑
4_0.53415*10^
‑
4_0.83705*10^
‑
4_0.95735*10^
‑
5_0.2875*10^
‑
5_0.51105*10^
‑
5_0.81265*10^
‑
5_0.95
165*10^
‑
8_0.2205*10^
‑
8_0.5275*10^
‑
8_0.8325*10^
‑
8_0.95
[0079]
在步骤6)中,分成4步进行。
[0080]
步骤6)
‑
1:在将表型训练集合和基因训练集合按照样本编码进行合并,含有2个正常等位基因的snp视为该snp的风险等位基因的数量为0;将含有1个正常等位基因的snp视为该snp的风险等位基因的数量为1;将含有0个正常等位基因的snp视为该snp的风险等位基因的数量为2(保证与步骤4中的基因型编码方式一致,即保证正常和风险等位基因一致)。早发性白发编码按照“病例对照范式”进行编码,case编码为1,control编码为0。可使用开源统计软件r和第三方软件包snpstats中的read.plink()函数进行基因数据的读取和编码。
[0081]
步骤6)
‑
2:按上述p_t策略过滤后的snp位点集合抽取训练集合的相应的子集合,添加性别(gender)作为候选预测因子,男性编码为1,女性编码为2。
[0082]
步骤6)
‑
3:根据p_t策略中的r2和p值组合,自适应的选择机器学习常用的特征选择算法对位点集合再次进行过滤。使用三种常用的特征选择算法剔除可能无效位点、效应微小位点和具有共线性的冗余位点。自适应的基本原则为:p在5*10^
‑
5水平下续选择的算法为lasso,随机森林类的boruta和逐步回归step;p在5*10^
‑
8水平下选择的算法为lasso和逐步回归step,p在5*10^
‑
4水平下选择的算法仅为lasso。其中lasso使用r中的第三方软件包glmnet中的cv.glmnet函数(参数:family="binomial",type.measure="auc",standardize=true,nfold=5),将回归系数不为0的特征作为输出特征;其中随机森林类的boruta使用lasso使用r中的第三方软件包boruta中的boruta()、tentativeroughfix()和getselectedattributes()函数进行特征选择。逐步回归step直接使用内置的step(fit,direction="both")函数进行选择,fit为未过滤位点用glm函数拟合的逻辑回归模型。将最后逐步回归敲定的模型包含的特征作为输出特征集合。
[0083]
步骤6)
‑
4:使用每种(共计19种,特征选择算法与p_t策略的组合)组合下每种特征选择算法选择的特征,使用逻辑回归,建立早发性白发的二元逻辑回归模型,因变量为case和control组成的0/1编码的早发性白发状态,自变量为特征选择后的位点集合组成的基因训练数据集合。
[0084]
在步骤7)中,将18种模型分别运用到独立的验证集合中,取机器学习中常用的auc值作为评估模型在验证集合中的分类效能,如下表5所示。
[0085]
表5
[0086]
分类效能最优的模型为验证集合中auc值最高的模型。其中测试集合和验证集合进行附加过滤(不包含任何位点的缺失值,分别过滤217和108个样本),保证其验证和测试的准确性。选择的p_t组合为:p=5*10^
‑
8,r2=0.95,选择的特征选择方法:step",选择的特征个数为:17(16个snp+gender)。最优早发性白发遗传风险评估模型所涉及的分子标记和分子权重在上述发明内容部分中已展示。
[0087]
步骤8)中,为了验证最优模型在人群中的早发性白发预测和风险区分能力。将最优模型运用到独立的测试集合中,评估其预测性能。其auc:0.66
‑
0.68。
[0088]
在由早发性白发和非早发性白发人群组成的独立测试集合中,case(早发性白发,右边灰色)和control(非早发性白发,左边黑色)群体的遗传风险得分分布的箱线图如图6所示。纵轴为基因风险分数,图中两个分组中的小圆点代表了该组基因风险分数的平均值。可以看出在测试集合中,早发性白发群体的遗传风险整体高于非早发性白发群体的分数。
[0089]
由早发性白发和非早发性白发群体组成的独立测试集合的50分位数图如图7所示。其中,上图,在由早发性白发和非早发性白发样本,年龄在20
‑
60岁组成的集合中,模型的预测分数的50分位数和早发性白发占比的关系图,强调了整体和分性别的患早发性白发的分布情况。x轴为分位数,y轴为早发性白发样本比例;下图为模型在男性和女性单独集合中的早发性白发遗传风险分数的分位数与早发性白发占比分布图。
[0090]
在由早发性白发和非早发性白发样本,年龄在20
‑
60岁组成的集合中,模型的预测分数的20和50分位数和早发性白发占比的关系图如图8所示,强调了两边极端组的早发性
白发占比情况。x轴为分位数,y轴为早发性白发样本比例。最低分位数组中早发性白发仅占3%;最高分位数组中早发性白发占据高达83%。
[0091]
如图9所示,由早发性白发和非早发性白发样本,年龄在20
‑
60岁组成的集合给出的50分数下的实际绝对风险与基于双变量正态分布估计的50分位数内的绝对风险关系图。强调了模型区分能力的客观和准确性。x轴为预测的每个分位数的早发性白发流行率,y轴为预测的本发明的独立集合中每个分位数的实际早发性白发流行率。
[0092]
在100分位数的分组中,最高百分位数和中间分位数之间的case/control的比值比(or)为11.66,相对风险比值(rr)为2.37;最高(top 1%)百分位数和最低分位数(bottom 1%)之间的case/control的比值比(or)为79.33,相对风险比值(rr)为11.04。在选择分类阈值为0.5时,敏感度(sensitivity)为0.89,阳性正确率(ppv)为0.67;整体准确性(accuracy):为0.66。提高分类阈值线,可提高早发性白发群体的检测敏感性和阳性正确率。
[0093]
为验证该模型对早发性白发的严重程度的预测能力。我们对在问题“你是否有过早发性白发”为肯定回答的,且根据问题“在最严重的时候,你的早发性白发情况最接近哪一种?”的回答选项,依次把回答为“稍微有些白发”、“接近一半白发”、“大部分头发变白”的人群作为案例组(case)编码为1;在问题“你是否有过早发性白发”为否定回答的作为非早发性白发群体即对照组(control)编码成0。
[0094]
结果显示出:该模型对早发性白发的严重程度的人群也具有预测区分能力:对“稍微有些白发”人群,其auc为0.65;对“接近一半白发”人群,其auc为0.69;对“大部分头发变白”人群,其auc为0.69。
[0095]
实施例2:构建一种早发性白发遗传风险基因检测试剂盒
[0096]
有关试剂盒的位点和引物设计流程:
[0097]
根据实施例1得到早发性白发遗传风险预测模型,选取其中的snp位点作为试剂盒的基因分子位点,形成检测panel模型。针对建立的早发性白发遗传风险水平检测panel模型涉及的所有snp位点,进行多重pcr扩增上下游引物对和单碱基延伸引物序列(如发明内容所示)的设计,引物设计示意图如图10所示。
[0098]
有关试剂盒的详情说明
[0099]
1.1主要组成成分
[0100]
pcr反应混合液,taq酶,扩增引物混合液,sap buffer,sap enzyme,单碱基延伸反应混合液,iplex enzyme,延伸引物混合液,ddh2o,阳性对照品,脱盐树脂,质谱芯片。
[0101]
1.2储存条件
[0102]
本品保存于
‑
20℃。
[0103]
1.3配套仪器
[0104]
通用pcr仪;dr massarray。
[0105]
1.4样本要求
[0106]
本品适用于从口腔黏膜细胞、口腔脱落细胞、唾液、血液和干血片中提取的基因组dna,要求dna a260/a280比值应介于1.8到2.0之间。冷冻dna样本应在
‑
20℃以下,并且避免反复冻融。
[0107]
1.5检验方法
[0108]
一、pcr反应
[0109]
1)按表6依次加入相关试剂组分配置5ulpcr反应体系,并分装至96孔pcr板中,3ul/孔。
[0110]
表6表6
[0111]
2)取出dna模板,冰上(4℃)熔解,涡旋震荡10s后简短离心,吸出一定量dna稀释至5ng/μl,备用。
[0112]
3)向96孔板中每孔加入2μl 5ng/μl dna模板,盖上管盖,涡旋震荡10s后简短离心备用,每次实验时必须设定空白对照(2μl ddh2o)、阴性对照(2μl dna提取洗脱液)和阳性对照。
[0113]
4)将96孔pcr板放入扩增仪,运行程序:pcr,具体程序如下:
[0114]
完成后,置于4℃保温。
[0115]
二、sap反应
[0116]
pcr反应结束后,在1.5ml ep管中配制sap混液。sap反应混合液成分包括:sap缓冲液
ꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀ
0.17μl cutsmart buffer(生产商neb)sap酶
ꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀ
0.5u(产商neb)ddh2o
ꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀ
补齐至2μl。
[0117]
1)每一个孔里加2μl sap混液(加混液后总体积:7μl)。
[0118]
2)把板用膜(用life的或其它公司质量较好的膜)封好,涡旋震荡和离心(4000rpm,5秒)。
[0119]
3)将板放上pcr仪进行以下程序:37℃
ꢀꢀꢀꢀꢀꢀꢀꢀꢀ
40分钟,85℃
ꢀꢀꢀꢀꢀꢀꢀꢀꢀ
5分钟,4℃保温。
[0120]
三、延伸反应
[0121]
1)取出sap反应板2000rpm离心1min。
[0122]
2)根据下表7在1.5ml管中配制iplex延伸混液。表7的数字是以一块96孔板加上
38%过剩额计算出来的。请根据实际反应的数量调整数字。
[0123]
表7单碱基延伸反应混合液(由缓冲液+acyntps混合)0.4μl(产自neb)iplex enzyme1u(产自neb)延伸引物混合液0.94μl水补齐至2μl
[0124]
3)每一个孔加入2μl iplex延伸混液并混匀(加混液后总体积:9μl)。
[0125]
4)把板用膜封好,涡旋震荡和离心(4000rpm 5秒)。
[0126]
5)将96孔板放上pcr仪进行以下热循环:
[0127]
四、树脂纯化
[0128]
1)把洁净树脂(resin)铺平在96/15mg凹坑板(dimple plate)上,用刮板刮反复左右推平树脂,压实,使每孔树脂含量均匀,待树脂由深黄色变为浅黄色方可使用。
[0129]
2)在样本板的每一个有样本的孔里加入41μl水,封膜(用普通的膜即可),然后离心。
[0130]
3)轻轻将样本板凌空反转,放在已放树脂的凹坑板上,两块板的孔要一一对应。然后将凹坑板连样本板一起反转(过程中两块板不可水平移动),让树脂掉到孔里。
[0131]
4)用膜(用普通的膜即可)把板封好,放在旋转器上颠倒摇匀15分钟。
[0132]
5)以3200g(标准板离心机的4000rpm)将板离心5分钟。
[0133]
五、质谱分析
[0134]
使用dr massarray进行质谱分析。
[0135]
1)打开“板管理系统”软件,编辑实验计划文件,包括样本的位置,样本名称和所用的引物,连接质谱仪与所建立的实验计划文件。
[0136]
2)点start all图标,启动软件,并检查各项指示灯是否正常。
[0137]
3)点击“芯片托盘进入/退出”按钮,把芯片放在托盘上,再放到芯片甲板上,记录芯片的位置(左边记为1,右边记为2)。手不要触及芯片表面;将96板放到标有mtp1/2的位置,按a1在左下角的方向放,固定好;芯片第一次使用时,在校准品加样槽内加入75μl的校正标准品,芯片非第一次使用时不需添加校正标准品。然后再点击“芯片托盘进入/退出”按钮,关闭夹板。
[0138]
4)点击“添加/维持树脂”按钮,打开树脂槽,添加树脂或者补充高压灭菌纯化水。a.仪器第一次开机时需加28g树脂进入树脂槽内并加16ml灭菌纯化水混匀。b.第一次使用时把9g树脂全部倒入树脂槽内,并加入5.2ml高压灭菌纯化水,用枪头混匀。c.非第一次使用时,需观察液面,若水的液面低于树脂面,则应补充适量的高压灭菌纯化水,使得水的液面高于树脂面。d.树脂溶液加入树脂槽后,需尽快使用,且不能放置超过30天。
buffer mld。
[0150]
3)转移500μl唾液样本至离心管,颠倒混匀3次,高速涡旋混匀15sec。放入45℃恒温孵育箱10min。
[0151]
4)将离心管放置于磁力架上静置30sec,待磁珠完全吸附后小心去除液体。
[0152]
5)将离心管从磁力架上取下,加入600μl buffer rbp,涡旋15sec重悬磁珠。
[0153]
6)将离心管放置于磁力架上静置30sec,待磁珠完全吸附时用移液枪小心去除液体。
[0154]
7)重复操作步骤6)和7)一次。
[0155]
8)将离心管从磁力架上取下,加入600μl 80%乙醇,涡旋15sec重悬磁珠。
[0156]
9)将离心管放置于磁力架上静置30sec,待磁珠完全吸附后用移液枪去除液体。
[0157]
10)将离心管从磁力架上取下,加入600μl无水乙醇,涡旋15sec重悬磁珠。
[0158]
11)将离心管放置于磁力架上静置30sec,待磁珠完全吸附时用移液枪小心去除液体。
[0159]
12)将离心管放置于磁力架上,打开管盖,室温晾干10min。
[0160]
13)将离心管从磁力架上取下,加入50μl buffer eb,用移液枪吸打混匀后,70℃孵育5min。
[0161]
14)短暂离心后将离心管放置于磁力架上静置1
‑
2min,直至磁珠被完全吸附。磁珠完全吸附后,将dna溶液转移至一个新的做好标记的0.6ml离心管中。
[0162]
将提取的dna,按照实施例2提供的实验步骤,使用早发性白发基因检测试剂盒和飞行时间质谱仪、typer4.0软件分型并输出检测结果。
[0163]
mofang
‑
hignmyopia_01的基因分型结果如下表9所示:
[0164]
表9
[0165]
1.2通过早发性白发遗传风险检测试剂盒对受检者(男性)进行snp基因分型后,获取检测样本的性别信息,按照模型里明示的每个snp位点的风险等位基因和该位点的基因检测结果,根据风险等位基因的个数进行编码(正常纯合为0,杂合为1,风险突变纯合为2),性别男编码为1,性别女编码为2,编码后的结果在上表第三列所示。
[0166]
计算公式:早发性白发遗传风险分数=snp_1*coffi_1+snp_2*coffi_2+.....+snp_16*coffi_16+gender*coffi_gender+截距*1
[0167]
如上公示所示,特征编码后的值乘上模型中对应的权重值,然后进行求和,加上截距项(
‑
1.25867205),所得分数即为对受检者的早发性白发遗传风险分数。
[0168]
1.3按照风险划分体系对所得分数进行风险评级。通过模型对基因数据和性别信息进行整合,我们可以得到其遗传风险分数为0.7761105。配套了早发性白发风险划分体系,按照权利要求7中给定的分割区间进行风险等级划分。
[0169]
划分结果为:mofang
‑
earlygreyhair_01的个人早发性白发基因风险为最高“等级6”,风险程度为“极高风险人群”,明显高于早发性白发流行率(32%)。每个风险等级的人群占比分布如下表10所示。
[0170]
表10风险等级该等级人群占比等级110.9%等级218.3%等级323.8%等级422.7%等级517.0%等级67.3%
[0171]
处于等级6的人群中,根据具有大样本量的独立测试集合的数据分布显示,该等级中实际的早发性白发vs非早发性白发人群比例为:67.2%vs 32.8%。
[0172]
以上所述的,仅为本发明的较佳实施例,并非用以限定本发明的范围,本发明的上述实施例还可以做出各种变化。凡是依据本发明申请的权利要求书及说明书内容所作的简单、等效变化与修饰,皆落入本发明专利的权利要求保护范围。本发明未详尽描述的均为常规技术内容。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1