一种(S)-1-(4-氯苯基)-1,3-丙二醇的酶法生产方法与流程
一种(s)
‑1‑
(4
‑
氯苯基)
‑
1,3
‑
丙二醇的酶法生产方法
技术领域
1.本发明属于有机合成领域,尤其涉及一种(s)
‑1‑
(4
‑
氯苯基)
‑
1,3
‑
丙二醇的酶法生产方法。
背景技术:
2.(s)
‑1‑
(4
‑
氯苯基)
‑
1,3
‑
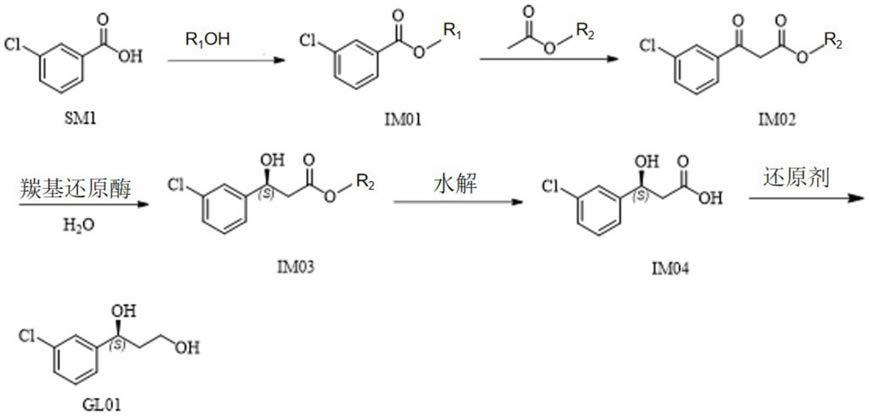
丙二醇是靶向肝脏药物所使用的关键中间体,已在多个临床及临床前研究药物中使用。例如甲磺酸帕拉德福韦,该肝靶向前药目前由西安新通医药股份有限公司开发,已完成临床二期,目前正在开展临床三期。该关键双醇中间体均采用化学的合成路线。
[0003][0004]
在专利文献us20030225277中报道了制备方法,工艺路线如下:
[0005][0006]
us2003/229225,以及文献jacs,2004,vol 126,#16,p5154
‑
5163也报道了类似的合成路线。
[0007]
上述合成路线在规模化生产时存在三点限制:
[0008]
(1)需要低温的操作条件,特别是第二步反应需要超低温条件
‑
60℃,对生产设备要求较高,规模化生产有难度;
[0009]
(2)需要使用有机催化剂(+)
‑
dipcl(cas no:112246
‑
73
‑
8),该催化剂价格昂贵,催化工艺操作繁琐,难以应用于规模化生产。
[0010]
(3)还原羧酸使用硼烷,该步反应规模生产的成本较高,且淬灭反应剧烈放热,具有一定安全风险。
[0011]
(4)制备得到的双醇化合物1的手性纯度为98%,为了满足注册申报要求,需要对其进行精制,进一步增加了生产成本。
[0012]
上述4点缺陷导致产品的生产成本较高,难以满足规模化生产的需要。因此迫切需
要一种可以规模化生产高光学纯度的(s)
‑1‑
(4
‑
氯苯基)
‑
1,3
‑
丙二醇规模化的方法。
技术实现要素:
[0013]
为了克服现有技术中制备关键医药中间体(s)
‑1‑
(4
‑
氯苯基)
‑
1,3
‑
丙二醇制备过程成本高,光学纯度低的缺陷,本发明利用特定羰基还原酶及其突变体作为催化活性物质,制备得到了关键的高立体选择性的手性醇化合物(im04),手性纯度>99.4%,再继续还原得到目标产物(s)
‑1‑
(4
‑
氯苯基)
‑
1,3
‑
丙二醇,本发明方法不需要要苛刻的低温条件,利于大规模的产业化生产。
[0014]
为解决上述技术问题,本发明提供了以下的技术方案:
[0015]
一种(s)
‑1‑
(4
‑
氯苯基)
‑
1,3
‑
丙二醇的酶法生产方法,合成路线如下:
[0016][0017]
其中,r1,r2独立地选自c1
‑
6的烷基,比如甲基,乙基,丙基,丁基。
[0018]
所述羰基还原酶具有对应于seq id no:2的氨基酸序列,或者对应于对应于seq id no:2的氨基酸序列存在下述突变之一或者两种以上的组合:45位的v突变为i(v45i),63位的k突变为q(k63q),141位的g突变为a(g141a),141位的g突变为v(g141v),195位的i突变为l(i195l),204位的a突变为v(a204v)。
[0019]
seq id no:2的氨基酸序列对应的羰基还原酶,命名wo03,来源于novosphingobium aromaticivorans的羰基还原酶nakred,参考文献cn102482648,发明人通过酶库的筛选,预料不到地发现wo03羰基还原酶的特定位点突变为特定氨基酸后得到的突变体,在以im02作为底物制备im03时,酶活性高,转化率高,立体选择型好。
[0020]
优选地,所述羰基还原酶突变体,是在对应于seq id no:2的氨基酸序列存在下述突变之一:
[0021]
(i)141位的g突变为a(g141a),且195位的i突变为l(i195l),其氨基酸序列如seq id no:4所示;
[0022]
(ii)141位的g突变为v(g141v),且195位的i突变为l(i195l),其氨基酸序列如seq id no:6所示。
[0023]
(iii)45位的v突变为i(v45i);141位的g突变为v(g141v,i195l),195位的i突变为l(i195l),其氨基酸序列如seq id no:8所示;
[0024]
(iv)45位的v突变为i(v45i),141位的g突变为v(g141v,i195l),195位的i突变为l(i195l),204位的a突变为v(a204v),其氨基酸序列如seq id no:10所示;
[0025]
(v)45位的v突变为i(v45i),63位的k突变为q(k63q),118位的l突变为m(l118m),141位的g突变为v(g141v,i195l),204位的a突变为v(a204v),其氨基酸序列如seq id no:12所示。
[0026]
所述羰基还原酶突变体还包括以下范围:对seq id no:4、6、8、10、或12所示的氨基酸序列在保持酶活性范围内,进行一个或多个氨基酸的替换、缺失、改变、插入或增加,所得到的氨基酸序列;或
[0027]
对seq id no:4、6、8、10、或12所示的氨基酸序列在保持酶活性范围内,在序列的n端或c端进行一个或多个氨基酸的插入,所述插入的氨基酸残基个数为1
‑
20个;优选1
‑
10个,更优选1
‑
5个。
[0028]
所述羰基还原酶的制备是将实现辅酶再生的醇脱氢酶或葡萄糖脱氢酶或甲酸脱氢酶与目的基因构建在同一质粒pet28a(+)载体上,然后导入表达宿主大肠杆菌,通过诱导表达,获得含有目的酶的菌体。可直接使用离心获得菌体,将其破壁获得粗酶液或粗酶粉。
[0029]
进一步地,本发明提供的(s)
‑1‑
(4
‑
氯苯基)
‑
1,3
‑
丙二醇的酶法生产方法,包括以下步骤:
[0030]
(s1)以间氯苯甲酸和醇r1oh酯化反应得到间氯苯甲酸烷基酯im01;
[0031]
(s2)间氯苯甲酸烷基酯(im01)在有机碱作用下和乙酸烷基酯ch3coor2缩合,得到产物酮酸酯im02;
[0032]
(s3)以酮酸酯im02为酶催化反应的底物,在羰基还原酶催化下,制备得到产物im03;
[0033]
(s4)im03经过水解得到im04;
[0034]
(s5)im04还原得到最终产物gl01,(s)
‑1‑
(4
‑
氯苯基)
‑
1,3
‑
丙二醇。
[0035]
优选地,步骤(s1)中酯化反应的条件为本领域所熟知,比如在酸催化剂下,可选择的酸包括浓硫酸,对甲苯磺酸,三氟甲磺酸,磷钨酸,阳离子交换树脂等。反应温度60
‑
100℃,在加热回流条件下反应15
‑
20h。间氯苯甲酸和r1oh的比例没有特别限定,一般选择醇r1oh过量,醇即作为反应物,也作为反应体系的溶剂。在本发明一个具体实施例中,间氯苯甲酸和r1oh的的摩尔比为0.3
‑
0.6:1。
[0036]
步骤(s1)酯化反应后的后处理为本领域所熟知,一般反应结束后,蒸除过量的醇,加入低极性溶剂(正庚烷,正己烷,石油醚),再分别用水、弱碱、盐溶液萃取洗涤,浓缩低极性溶剂相得产物im01,产率在88
‑
93%。
[0037]
优选地,步骤(s2)中有机碱选自有机醇的钾盐和/或钠盐,比如叔戊醇钾,叔戊醇钠,叔丁醇钾,叔丁醇钠,甲醇钠,甲醇钾,乙醇钠,乙醇钾中的至少一种。im01,酯ch3coor2和有机碱的摩尔比为1:1.1
‑
1.7:1.5
‑
2.5,优选1:1.3
‑
1.5:1.8
‑
2.2。
[0038]
步骤(s2)反应温度
‑
5至5℃,反应时间1
‑
3h,反应溶剂为thf,丙酮,dmso,dmf,溶剂含水量<1000ppm。反应结束后加入酸猝灭反应,之后后处理为本领域所熟知,具体是加入酸淬灭反应后,静置分层,上层溶剂浓缩,下层水相用溶剂萃取,和上层浓缩液合并,分别依次经过水,弱碱,盐洗涤,浓缩得到产物im02,收率85
‑
90%。
[0039]
优选地,步骤(s3)酮酸酯im02作为酶催化反应的底物,浓度为50~200g/l;更优选
为100~150g/l。
[0040]
进一步地,所述羰基还原酶为游离形式的酶、固定化酶、或菌体形式的酶中的至少一种。羰基还原酶的用量是反应体系中底物的1wt%~6wt%。在本发明一个具体实施方式中,使用羰基还原酶的湿菌体,湿菌体质量与底物的质量比率为30wt%~100wt%,优选50wt%
‑
70wt%。
[0041]
进一步地,所述的反应体系中,还存在共底物,所述的共底物选自下组:异丙醇、葡萄糖、甲酸铵中的至少一种;优选地,共底物的浓度为底物浓度的100
‑
200%,优选共底物的浓度为底物浓度的140
‑
170%。
[0042]
进一步地,步骤(s3)反应过程中在磷酸缓冲盐体系中进行,ph为6
‑
8,较佳地6.5
‑
7.5,更佳地6.8
‑
7.1;和/或反应温度为20℃
‑
50℃,较佳地25℃
‑
40℃,更佳地30℃
‑
35℃;和/或反应时间3
‑
20小时,更佳地3
‑
10小时。
[0043]
进一步地,所述的辅酶指能够实现氧化还原反应中电子传递的辅酶;包括还原型辅酶、氧化型辅酶中的至少一种;所述还原型辅酶选自nadh、nadph、或其组合;所述氧化型辅酶选自nad
+
、nadp
+
或其组合;更进一步地,所述辅酶用量与底物用量比率为0.01wt%~1.0wt%、优选为0.1wt%~0.5wt%。
[0044]
进一步地,所述羰基还原酶和/或用于辅酶再生的酶的基因构建在表达载体上。
[0045]
进一步地,所述的反应体系中,还存在用于辅酶再生的酶,具体选自醇脱氢酶,甲酸脱氢酶、葡萄糖脱氢酶、或其组合。
[0046]
优选地,步骤(s4)所述水解是在碱存在的情况下进行,所述碱没有特别的限定,一般为无机碱,比如氢氧化钠和/或氢氧化钾,碱的用量没有特别的限定,能完全充分水解即可,一般是调节ph为12
‑
14。水解之后的后处理操作为本领域所熟知,具体是用酸调节ph为7
‑
9,加入有机溶剂萃取洗去有机杂质,调节ph至1
‑
2,静置分层,有机溶剂萃取,水洗,干燥,过滤,浓缩,得油状物。油状物用乙酸乙酯复溶,加热至35
‑
50℃,滴加低极性有机溶剂,并降温至
‑
5至5℃,析出大量固体,过滤,洗涤,干燥得针状固体,为产物im04。步骤(s4)收率在70
‑
80%,产物纯度在99%以上。
[0047]
优选地,步骤(s5)中,分批加入还原剂,所述还原剂为硼氢化钠和/或硼氢化钾,用量为im04质量的5
‑
10%,反应温度
‑
5℃至5℃,加入时间为1
‑
2h,还原剂加入完毕后,维持低温继续反应10
‑
30min,之后缓慢滴加三氟化硼乙醚,三氟化硼乙醚用量为im04的1
‑
2倍,在1
‑
3h内滴加完毕,维持低温反应1
‑
3h,之后升温至20
‑
30℃,反应3
‑
5h,再降温至10
‑
20℃后滴加甲醇,在1
‑
2h内滴加完毕继续反应1
‑
2h,之后进行后处理步骤即得产物gl01。所述后处理为本领域所熟知,在本发明一个具体实施方式中,后处理是反应结束后,调节体系为弱碱性,用有机溶剂萃取后,洗涤,减压浓缩,得到淡黄色粘稠液体,为最终产物gl01,即(s)
‑1‑
(4
‑
氯苯基)
‑
1,3
‑
丙二醇。
[0048]
本发明对羰基还原酶进行了筛选,得到了野生型的羰基还原酶wo03及其突变体,发现其对于关键医药中间体(s)
‑1‑
(4
‑
氯苯基)
‑
1,3
‑
丙二醇的手性还原具有很好的催化活性,酶活性高,立体选择型好,手性纯度>99.6%,优选实施例可以达到99.8%以上。本发明方法不需要要苛刻的低温条件,利于大规模的产业化生产。
附图说明
[0049]
图1是实施例2步骤(s3)所得产物im03的手性图谱;
[0050]
图2是实施例2步骤(s3)所得产物im03的1h nmr图谱;
[0051]
图3是实施例2步骤(s5)所得产物gl01的1h nmr图谱;
[0052]
图4是实施例2步骤(s5)所得产物gl01的
13
c nmr图谱;
[0053]
图5是实施例2步骤(s5)所得产物gl01的hplc化学纯度图谱;
[0054]
图6是实施例2步骤(s5)所得产物gl01的hplc手性纯度图谱。
具体实施方式
[0055]
制备例
[0056]
制备例羰基还原酶工程菌及羰基还原酶同源突变库的构建
[0057]
将来自wo03羰基还原酶酶基因及来自其突变体基因的5种羰基还原酶酶基因克隆入pet28a(+)载体,然后导入宿主大肠杆菌bl21,通过诱导表达,获得羰基还原酶的重组基因工程菌。
[0058]
以野生型的羰基还原酶wo03(其核酸序列如seq no.1所示,其氨基酸序列如seq no.2所示)为模板,利用随机点突变试剂盒(ii site
‑
directed mutagenesis kit)进行易错pcr,或通过定向进化将野生型的羰基还原酶基因进行突变,获得包含进化的羰基还原酶基因的质粒文库。构建后的质粒文库转入大肠杆菌bl21(de3)(货号:康为世纪cw0809s)中,于37℃烘箱中,在含50μg/ml卡那霉素的lb固体培养基上培养过夜。将单菌落挑选至含400μl lb液体培养基(含50μg/ml卡那霉素)的96孔板中,37℃,200rpm过夜培养,得种子液。然后将10μl种子液转移至含400μl发酵培养基(含50μg/ml卡那霉素)的96深孔板中,37℃培养至od600值>0.8。然后加入终浓度为1mm的异丙基硫代半乳糖苷(iptg),降温至28℃诱导突变体表达,继续培养20h。发酵结束后,通过4000g,30min离心收集菌体,然后再用200μl裂解缓冲液(含有1000u溶菌酶的0.1m磷酸缓冲液,ph 7.0)重新悬浮菌体,并在30℃裂解1h。裂解结束后,于4℃,4000g,离心30min,澄清的上清液用于测定突变体活性。将190μl反应液(含0.4mm底物、1mm nadh、40μl二甲基亚砜)加入新的96孔板中,再加入10μl的上清液后,在340nm检测nadh的变化,nadh的消耗量反应突变体酶活的高低,筛选得到如下表1所示的羰基还原酶突变体。
[0059]
表1
[0060][0061]
实施例1
[0062]
以化合物ⅳ为底物,对羰基还原酶进行筛选,筛选方法与筛选结果如下;
[0063][0064]
反应转化率的检测方法:phenomenex gemini c18 4.6*250mm 5μm);流动相a为10%乙腈,流动相b为乙腈,按下表进行梯度洗脱;流速为每分钟1.0ml;柱温为30℃;检测波长为220nm。
[0065][0066][0067]
化合物
ⅴ
的手性监测方法如下:色谱柱:大赛璐ib
‑
3,5μm,4.6*250mm;流动相:异丙醇∶正己烷=10:90;流速1.0ml/min;运行时间:20min;柱温30℃;检测波长:220nm。s构型10min,r构型13min。
[0068]
结果如下表2所示:
[0069]
表2
[0070]
酶编号转化率e.e.值构型wo03(seq id no.2)
[a]
98.9%99.80%slsadh
[b]
21.3%93.78%rlk
[b]
12.4%38.52%s
[0071]
注:羰基还原酶lsadh来源于leifsonia sp.strain s749,登记号为ab213459,羰基还原酶lk来源于lactobacillus kefir,参考文献wo 2010/025238。
[0072]
反应条件:(a)1g iv化合物,0.2g湿菌体,0.001g nadp+,0.1g gdh湿菌体,1.5g葡萄糖和10ml磷酸缓冲液(100mm,ph 7.0)于30℃、220rpm摇床反应24h;(b)1g iv化合物,0.2g湿菌体,0.001g nad+,20%异丙醇和10ml磷酸缓冲液(100mm,ph 7.0)于30℃、220rpm摇床反应24h。
[0073]
可以看出,上述羰基还原酶,除wo03(氨基酸序列如seq id no:2所示,其编码基因如seq id no:1所示)的转化率和立体选择性都不能满足工业化生产的要求,不适合大规模的生产关键中间体(s)
‑1‑
(4
‑
氯苯基)
‑
1,3
‑
丙二醇,但羰基还原酶wo03的酶活性还不够高,因此,本发明以wo03的羰基还原酶作为基础,对其进行突变,筛选出酶活性,转化率和立体选择性皆优异的羰基还原酶突变体。
[0074]
将制备例1中保存于甘油中的重组基因工程菌,接种到含100ug/ml氨茉霉素的lb液体培养基中,37c,220rpm,培养12
‑
16h,得到种子培养基,将此种子液按1.5%的比例接种到含100ug/ml氨茉抗性的液体培养基中,然后37℃,220rpm培养至od
600
值>2.0,加入终浓度为1.0%乳糖,降温至25c,继续培养2h,加入终浓度为0.5%的乳糖,培养20h,放罐,离心得菌体,用于生物转化的催化剂。按照实施例1的反应条件(a),即:0.1g iv化合物,0.2g湿菌体,0.001g nadp+,0.1g gdh湿菌体,0.2g葡萄糖和10ml磷酸缓冲液(100mm,ph 7.0)于30℃、220rpm摇床反应24h制备化合物v。对所得羰基还原酶同源突变库库进行筛选,结果如下表3所示:
[0075]
表3
[0076][0077]
酶活性的定义:在50ml离心管内反应,称取底物底物0.5g,加入异丙醇1.5ml,加入
ph6.0
‑
6.5磷酸缓冲液3.5ml,称取辅酶nad 0.025g,将以上体系在30℃水浴锅预热15min称取0.5g菌体,加入反应体系,放入30℃恒温室摇床150rpm,开始计时1h。结束后,在摇匀的状态下快速用移液枪移去0.1ml反应液,加入10ml离心管中,然后加入4.9ml异丙醇,用移液枪充分混匀。用离心机离心10min,3500rpm。将上清液倒入取样管中,检测转化率,即为酶活性。
[0078][0079]
由于密码子的简并性,如上羰基还原酶突变体的核酸序列不局限于表1中所列出核酸序列。本领域技术人员可以通过适当引入替换、缺失、改变、插入或增加来获得该碱基序列的同系物,本发明涵盖这些同系物,只要其表达的重组酶保持对im02的化合物的催化还原活性即可。本发明中多聚核苷酸的同系物可以通过对碱基序列的一个或多个碱基在保持酶活性范围内进行替换、缺失或增加来制得。
[0080]
本发明提供了氨基酸序列如seq id no:4、6、8、10、或12所示的羰基还原酶突变体,其相对于野生型酶wo03均具有明显改善的酶活性,能够减少酶的用量,对s构型产物的立体选择性均可达到99%以上。因此,借助wo03羰基还原酶及其突变体,可以有效地将im02化合物还原为s构型的手性醇化合物im03,产物的收率,立体选择性,酶的利用效率都领人满意,有望借助特定酶的催化活性,制备关键中间体(s)
‑1‑
(4
‑
氯苯基)
‑
1,3
‑
丙二醇。
[0081]
实施例2
[0082]
(s1):
[0083][0084]
将1.5kg sm1投入20l反应釜,加入无水乙醇8.3kg,随后加入硫酸,升温,至80℃,保温18h,hplc检测,至95%产物,开始浓缩反应液,蒸除乙醇。浓缩物加4l正庚烷,再加入4l水洗,水洗后用4l饱和碳酸氢钠水溶液洗涤,再用4l饱和食盐水洗涤,浓缩正庚烷即得产物im01 1.6kg。
[0085]
(s2):
[0086][0087]
将thf 7.6kg(含水量<1000ppm)加至20l反应釜,设定温度为
‑
5℃,加入叔丁醇钾(99.5%)1.9kg,搅拌至溶清。随后加入1.6kg im01,温度为0℃以下即滴加无水ea 0.99kg,于90min滴毕,5℃保温2h,hplc监测。原料<1%,加入3n hcl淬灭。随后静止分层。上层浓缩thf,下层用2l ea萃取下层一次,与上层浓缩物合并。再用ea稀释。经水洗、饱和碳酸钠洗、饱和nacl洗后,浓缩得im021.67kg。
[0088]
(s3):
[0089][0090]
将im02 1.67kg加至1.6l的0.1mol/lpbs缓冲溶液中,加入2.2kg异丙醇和0.53kg wo03湿菌体,随后加入3.2gnad
+
,在35℃水浴中,搅拌条件下反应,反应期间每隔一段时间(0.5
‑
1h)测试体系ph值,用饱和碳酸钠调节体系ph为6.9
‑
7.2,反应15h后,hplc监控原料剩余<0.5%,停止反应,浓缩异丙醇后,加热至70℃,加入500g硅藻土,过滤,得滤液,乙酸乙酯乙酸乙酯萃取水层两次(1x2l),合并有机层,饱和氯化钠水洗,无水硫酸钠干燥,过滤浓缩得淡黄色油状产物im03 1.47kg,ee值99.9%,其手性图谱和1hnmr图谱分别如图1和图2所示。1hnmr和
13
c nmr具体数据为:(600mhz,chloroform
‑
d)δ7.39(s,1h),7.26(dq,j=15.9,7.7hz,3h),5.24
–
4.94(m,1h),4.18(q,j=7.1hz,2h),3.52(s,1h),2.82
–
2.52(m,2h),j=7.2hz,3h).
13
c nmr(151mhz,chloroform
‑
d)δ172.20,144.60,134.44,129.82,125.95,123.81,69.65,61.04,43.19,14.13.
[0091]
(s4):
[0092][0093]
在5l的四口瓶中,投入im03(1.47kg),降温至10~15℃,开始滴加碱水(300g氢氧化钠溶于3l水),2h滴毕,控制ph=13~14,继续反应3h,hplc检测,原料<0.5%,加入浓盐酸调ph至8
‑
9,加入乙酸乙酯1l萃取水层洗去有机杂质,浓盐酸调节ph=1,分层,乙酸乙酯萃取水层(1.5l
×
2),合并有机层,饱和食盐水洗涤(1.5l),无水硫酸钠(200g)干燥,过滤,浓缩得粘稠油状物(包夹固体)。向上述油状物中加入乙酸乙酯(900ml),加热至40℃,溶清,滴加3.6l石油醚,降温至0℃,析出大量固体,保温3h,过滤,冷的乙酸乙酯/石油醚(1:4)洗涤,干燥得针状固体im04,995g,纯度99.7%,,母液回收85.5g。[α]
d25
=
‑
17.3
°
(c 1.00,meoh).esi
‑
ms(m/z):199.0[m
‑
h]
‑
.1h nmr(400mhz,chloroform
‑
d)δ7.40(q,j=1.4,0.9hz,1h),7.35
–
7.22(m,3h),5.14(dd,j=8.4,4.4hz,1h),2.88
–
2.72(m,2h).
13
c nmr(101mhz,cdcl3)δ176.66,144.08,134.62,129.97,128.18,125.95,123.79,69.52,42.83。
[0094]
(s5):
[0095][0096]
四口反应瓶、氮气保护,加入im04 20克、四氢呋喃160克,打开搅拌降温至
‑
5℃,控温
‑
5℃至0℃分批加入硼氢化钠6.8克,用时1
‑
1.5小时,在分批加入过程中,有明显放热现
象,注意温度变化,同时硼氢化钠较易吸潮,注意保护,在此温度下控温0℃以下反应20分钟,控温
‑
5—0℃,2.5小时左右滴加三氟化硼乙醚34克,在滴完后,
‑
3—0℃控温反应1小时后,再升温至5℃控温反应1小时,25℃控温反应4小时,降温至15—20℃滴加甲醇24克,用时1—1.5小时,滴加过程中,有明显放热现象,需要注意控温,观察反应液冒泡现象,控温反应1小时。30℃以下减压浓缩至干,加入水250搅拌溶解,控温10
‑
12℃滴加10%氢氧化钠溶液50克左右调ph值8.5—9.0,用时1.5左右,搅拌40分钟,再复测ph值不再有变化,控温12—15℃加入二氯甲烷150克,搅拌40分钟,静止分层40分钟分出下层有机相、用60克、40克再萃取两次,合并有机相,有机相加入饱和食盐水50克,用6n盐酸调至中性,搅拌1小时,静止40分钟,分层,30℃以下减压浓缩至不再有液体外出,再用油泵减压浓缩,得淡黄色粘稠液体15.5g,hplc化学纯度为99.4%,ee值99.8%。hrms m/z calcd for c9h
11
clnao2:209.0340,found 209.0347[m+na]
+
.1h nmr(600mhz,chloroform
‑
d)δ7.38(d,j=2.1hz,1h),7.33
–
7.18(m,3h),4.94(dd,j=8.6,3.8hz,1h),3.86(dt,j=7.2,4.2hz,2h),3.21(s,1h),1.95(dddd,j=20.3,16.1,8.7,4.7hz,2h).
13
c nmr(151mhz,chloroform
‑
d)δ146.43,134.42,129.79,127.62,125.89,123.79,73.66,61.36,40.33。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1