一种吲哚啉异色满类衍生物及其酶催化合成方法和应用
1.本发明属于精细化工领域,具体涉及一种吲哚啉异色满类衍生物及其酶催化合成方法和应用。
背景技术:
2.吲哚啉异色满类化合物是一类具有单萜类生物碱的结构,广泛存在于许多天然产物以及药物分子中,例如双多叶碱,钩藤碱中,现有研究证明其有潜在的生物活性,包括抗菌、抗炎和抗肿瘤作用。在有机化学界,双多叶碱的合成被认为是一个值得攀登的山峰。已有的文献报道吲哚啉异色满类化合物的合成往往需要过渡金属的运用,这在后面药物研究中特别是金属残有量中带来巨大的影响。因此,传统的化学合成吲哚啉异色满类化合物在化工生产上存在不实用,不经济,不环保的问题,开发一种绿色高效合成吲哚啉异色满类化合物是迫切需要的。
技术实现要素:
3.针对现有技术中的问题,本发明提供一种吲哚啉异色满类衍生物及其制备方法,解决现有技术操作复杂,生产成本高等缺点,从廉价易得的化学原料起,通过多铜氧化酶为催化剂催化的氧化偶联反应得到高附加值的吲哚啉异色满类化合物,且反应条件温和,操作简单,成本低廉,绿色环保。
4.为实现以上技术目的,本发明的技术方案是:
5.一种吲哚啉异色满类衍生物,其结构如下:
[0006][0007]
所述r1为氢、烷基、杂芳香基中的任意一种;r2为氢、卤素、烷基、烷氧基、硝基、酯基、氰基、酰基中任意一种;r3为氢、卤素、烷基、烷氧基、硝基、酯基、氰基、酰基、氨基和芳基中的任意一种。
[0008]
一种吲哚啉异色满类衍生物的制备方法,以2,3
‑
二羟基苯甲酸和吲哚类化合物为底物,多铜氧化酶为催化剂,反应得到吲哚啉异色满类衍生物,反应路线如下所示:
[0009][0010]
所述r1为氢、烷基、杂芳香基中的任意一种;r2为氢、卤素、烷基、烷氧基、硝基、酯
基、氰基、酰基中任意一种;r3为氢、卤素、烷基、烷氧基、硝基、酯基、氰基、酰基、氨基和芳基中的任意一种。
[0011]
所述吲哚类化合物为式(1)
‑
式(6)中的一种,且式(1)
‑
式(6)的结构如下:式(6)的结构如下:
[0012]
所述制备方法包括如下步骤:
[0013]
(1)将2,3
‑
二羟基苯甲酸、吲哚类化合物、多铜氧化酶依次加入至有机溶剂、水和酸的混合体系中进行催化反应;
[0014]
(2)反应结束后萃取分离,干燥后即可获得吲哚啉异色满类衍生物。
[0015]
其中,所述有机溶剂为二甲基亚砜、n,n
‑
二甲基甲酰胺和乙腈中的一种,优选为乙腈。
[0016]
所述酸采用盐酸、甲酸、乙酸中的一种,优选为盐酸。
[0017]
所述有机溶剂在混合体系中的体积占比为10
‑
50%,优选为50%。
[0018]
所述多铜氧化酶为细菌锰氧化酶、铜外排氧化酶、地衣芽孢杆菌漆酶中的至少一种;进一步的,所述多铜氧化酶采用地衣芽孢杆菌漆酶,所述地衣芽孢杆菌漆酶通过大肠杆菌发酵获得。
[0019]
所述多铜氧化酶与吲哚类化合物的物质的量比为0.01
‑
0.1:1,所述2,3
‑
二羟基苯甲酸与吲哚类化合物的物质的量比为1:1
‑
1.2,。
[0020]
所述制备方法内还包括辅助氧化剂,且所述辅助氧化剂与多铜氧化酶同时加入,所述辅助氧化剂为空气、30%过氧化氢水溶液和叔丁基过氧化氢水溶液tbhp中的至少一种,其中优选为30%过氧化氢水溶液。
[0021]
所述催化反应的温度为4
‑
37℃。
[0022]
上述的吲哚啉异色满类衍生物在制备药物或天然产物中的应用。
[0023]
从以上描述可以看出,本发明具备以下优点:
[0024]
1.本发明解决现有技术操作复杂,生产成本高等缺点,从廉价易得的化学原料起,通过多铜氧化酶为催化剂催化的氧化偶联反应得到高附加值的吲哚啉异色满类化合物。本发明的反应机理是通过地衣芽孢杆菌漆酶cota对2,3
‑
二羟基苯甲酸底物的氧化形成苯醌类的中间体,然后吲哚类化合物对该中间体的亲核加成,然后环化即可得到吲哚啉异色满类化合物。多铜氧化酶是一种含有金属铜中心的氧化还原酶,广泛存在于细菌、真菌和高等植物中。以研究最多的漆酶为例,通常由四个铜原子组成,根据三个活性位点进行分类:1型或蓝色位点、2型或正常位点、3型或双核位点。t1 cu活性位点接受底物氧化的4个电子并将它们传递给t2/t3 cu簇,在t2/t3 cu簇中,分子氧通过接受4个电子完全还原为2个水分子。
[0025]
2.本发明制备方法反应温和,一定比例的有机溶剂有利于反应底物的溶解,酸可以提高反应的转化率,成本低廉,操作简单,绿色环保。
[0026]
3.本发明制备方法转化率和收率高,所采用的地衣芽孢杆菌漆酶cota是通过大肠杆菌发酵而来,生产成本低于现有的技术,有利于工业化生产。
附图说明
[0027]
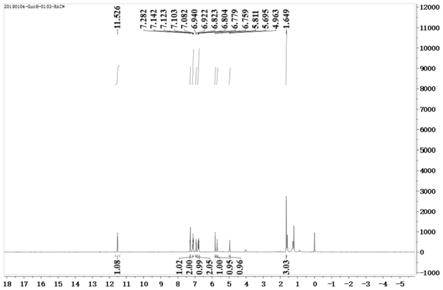
图1是实施例1制备的目标产物的核磁共振氢谱。
具体实施方式
[0028]
结合图1,详细说明本发明的一个具体实施例,但不对本发明的权利要求做任何限定。
[0029]
实施例中的2,3
‑
二羟基苯甲酸,双氧水,吲哚类化合物,盐酸均是购买自上海,所述地衣芽孢杆菌漆酶cota(ncbi accession no.yp_077905)是通过大肠杆菌发酵而来,购买至苏州金唯智生物科技有限公司。
[0030]
实施例1
[0031]
向一支10毫升的反应瓶中依次加入2,3
‑
二羟基苯甲酸(0.3mmol,46.2mg),3
‑
甲基吲哚(0.15mmol,19.7mg),cota(0.1mol%),双氧水(0.75mmol,75ul),去离子水(1ml),乙腈(1ml),盐酸(0.3mmol,13ul)室温下磁力搅拌48小时,待底物转化完后向反应体系中加入乙酸乙酯萃取(3ml*4),合并有机相并用无水硫酸钠干燥,过滤后真空浓缩得粗产品。所得粗产品通过柱层析法进行纯化分离,得到黄色固体产物,分离产率为87%。
[0032]
实施例1的合成路线如下所示:
[0033][0034]
图1为实施例1所得目标产物的核磁共振氢谱。
[0035]1h nmr(400mhz,cdcl3):11.52(s,1h),7.28(s,1h),7.11(q,j=15.6hz,8.0hz,2h),6.93(d,j=7.2hz,1h),6.79(q,j=17.6hz,8.0hz,2h),5.81(s,1h),5.69(s,1h),4.96(s,1h),1.64(s,3h)。
[0036]
实施例2
[0037]
向一支10毫升的反应瓶中依次加入2,3
‑
二羟基苯甲酸(0.3mmol,46.2mg),6
‑
氟
‑3‑
甲基吲哚(0.15mmol,22.4mg),cota(0.1mol%),双氧水(0.75mmol,75ul),去离子水(1ml),乙腈(1ml),盐酸(0.3mmol,13ul)室温下磁力搅拌48小时,待底物转化完后向反应体系中加入乙酸乙酯萃取(3ml*4),合并有机相并用无水硫酸钠干燥,过滤后真空浓缩得粗产品。所得粗产品通过柱层析法进行纯化分离,得到黄色固体产物,分离产率为85%。
[0038]
实施例2的合成路线如下所示:
[0039][0040]
所得目标产物的核磁共振氢谱如下:
[0041]1h nmr(400mhz,cdcl3):11.50(s,1h),7.30(s,1h),7.08(d,j=8.0hz,1h),6.85(q,j=5.6hz,8.8hz,1h),6.50(d,j=5.2hz,2h),5.83(s,1h),5.08(s,1h),1.65(s,3h)。
[0042]
实施例3
[0043]
向一支10毫升的反应瓶中依次加入2,3
‑
二羟基苯甲酸(0.3mmol,46.2mg),5
‑
氯
‑3‑
甲基吲哚(0.15mmol,24.8mg),cota(0.1mol%),双氧水(0.75mmol,75ul),去离子水(1ml),乙腈(1ml),盐酸(0.3mmol,13ul)室温下磁力搅拌48小时,待底物转化完后向反应体系中加入乙酸乙酯萃取(3ml*4),合并有机相并用无水硫酸钠干燥,过滤后真空浓缩得粗产品。所得粗产品通过柱层析法进行纯化分离,得到黄色固体产物,分离产率为83%。
[0044]
实施例3的合成路线如下所示:
[0045][0046]
所得目标产物的核磁共振氢谱如下:
[0047]1h nmr(400mhz,cdcl3):11.51(s,1h),7.31(d,j=8.4hz,1h),7.11
–
7.08(m,2h),6.88(d,j=2.0hz,1h),6.71(d,j=8.4hz,1h),5.82(s,1h),4.99(s,1h),1.66(s,3h)。
[0048]
实施例4
[0049]
向一支10毫升的反应瓶中依次加入2,3
‑
二羟基苯甲酸(0.3mmol,46.2mg),5
‑
溴
‑3‑
甲基吲哚(0.15mmol,31.5mg),cota(0.1mol%),双氧水(0.75mmol,75ul),去离子水(1ml),乙腈(1ml),盐酸(0.3mmol,13ul)室温下磁力搅拌48小时,待底物转化完后向反应体系中加入乙酸乙酯萃取(3ml*4),合并有机相并用无水硫酸钠干燥,过滤后真空浓缩得粗产品。所得粗产品通过柱层析法进行纯化分离,得到黄色固体产物,分离产率为64%。
[0050]
实施例4的合成路线如下所示:
[0051][0052]
所得目标产物的核磁共振氢谱如下:
[0053]1h nmr(400mhz,cdcl3):11.50(s,1h),7.31(d,j=8.4hz,1h),,7.25(dd,j=6.0hz,8.0hz,1h),7.08(d,j=8.0hz,1h),7.01(d,j=2.0hz,1h),6.67(d,j=8.4hz,1h),5.80(s,1h),5.00(s,1h),1.66(s,3h)。
[0054]
实施例5
[0055]
向一支10毫升的反应瓶中依次加入2,3
‑
二羟基苯甲酸(0.3mmol,46.2mg),3
‑
乙基吲哚(0.15mmol,21.8mg),cota(0.1mol%)),双氧水(0.75mmol,75ul),去离子水(1ml),乙腈(1ml),盐酸(0.3mmol,13ul)室温下磁力搅拌48小时,待底物转化完后向反应体系中加入乙酸乙酯萃取(3ml*4),合并有机相并用无水硫酸钠干燥,过滤后真空浓缩得粗产品。所得粗产品通过柱层析法进行纯化分离,得到黄色固体产物,分离产率为76%。
[0056]
实施例5的合成路线如下所示:
[0057][0058]
所得目标产物的核磁共振氢谱如下:
[0059]1h nmr(400mhz,cdcl3):11.56(s,1h),7.29(d,j=6.4hz,1h),7.15
‑
7.11(m,1h),7.02(t,j=8.4hz,2h),6.85
–
6.81(m,1h),6.76(d,j=8.0hz,1h),5.99(s,1h),4.97(s,1h),2.51
‑
2.15(m,1h),1.97
‑
1.88(m,1h),0.93(t,j=7.6hz,3h)。
[0060]
实施例6
[0061]
向一支10毫升的反应瓶中依次加入2,3
‑
二羟基苯甲酸(0.3mmol,46.2mg),2,3
‑
二甲基吲哚(0.15mmol,21.8mg),cota(0.1mol%),双氧水(0.75mmol,75ul),去离子水(1ml),乙腈(1ml),盐酸(0.3mmol,13ul)室温下磁力搅拌48小时,待底物转化完后向反应体系中加入乙酸乙酯萃取(3ml*4),合并有机相并用无水硫酸钠干燥,过滤后真空浓缩得粗产品。所得粗产品通过柱层析法进行纯化分离,得到黄色固体产物,分离产率为73%。
[0062]
实施例6的合成路线如下所示:
[0063][0064]
所得目标产物的核磁共振氢谱如下:
[0065]1h nmr(400mhz,cdcl3):11.57(s,1h),7.29(d,j=5.6hz,1h),7.12(d,j=8.4hz,2h),6.81(d,j=4.0hz,2h),6.76(d,j=7.6hz,1h),5.71(s,1h),4.93(s1h),1.76(s,3h),1.59(s,3h)。
[0066]
可以理解的是,以上关于本发明的具体描述,仅用于说明本发明而并非受限于本发明实施例所描述的技术方案。本领域的普通技术人员应当理解,仍然可以对本发明进行修改或等同替换,以达到相同的技术效果;只要满足使用需要,都在本发明的保护范围之内。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1