一种4-氯-2-氰基苯磺酰氯的合成方法与流程
一种4
‑
氯
‑2‑
氰基苯磺酰氯的合成方法
技术领域
1.本发明属于有机合成技术领域,具体涉及一种4
‑
氯
‑2‑
氰基苯磺酰氯的合成方法。
背景技术:
2.文献[cn201880075992.0]报道发明人已经发现了一系列具有优异的核糖核苷酸还原酶抑制活性的磺酰胺化合物,这些磺酰胺化合物作为结晶形式诸如共晶和/或作为盐形式,以低静电的且非吸湿性的形式存在,性质表现稳定。这些磺酰胺化合物是核糖核苷酸还原酶有效力的且选择性的抑制剂,可以在治疗与核糖核苷酸还原酶相关的其他疾病中用作抗肿瘤剂或治疗剂,4
‑
氯
‑2‑
氰基苯磺酰氯是合成上述磺酰胺化合物的重要中间体。
[0003]
截至目前,该中间体没有相同合成文献报道,类似物质合成如cn104603118报道了4
‑
溴
‑2‑
氰基苯磺酰氯的合成,wo2013061977报道了4
‑
氟
‑2‑
氰基苯磺酰氯的合成,这两种公开的合成方法都是通过重氮化反应合成目标产物。重氮化反应是化工生产中危险反应类型的一种,容易发生安全生产事故,且在医药产品中,重氮化反应是引入基因毒性杂质的一种反应类型(例如重氮化反应能产生肼结构的杂质r
‑
n=n
‑
r,此结构是产生基因毒性的警示结构,医药产品需要重点关注),增大了药物研究过程的工作量,因此研究提供一种能避开重氮化反应合成4
‑
氯
‑2‑
氰基苯磺酰氯的安全工业化生产方法,显得尤为迫切。
技术实现要素:
[0004]
本发明的目的在于提供一种能避开重氮化反应合成4
‑
氯
‑2‑
氰基苯磺酰氯的方法。
[0005]
基于上述目的,本发明通过提供一种4
‑
氯
‑2‑
氰基苯磺酰氯的合成方法,该方法所用原材料简单,价格低廉,反应步骤短,生产周期短,操作简单,易于实现工业化生产。
[0006]
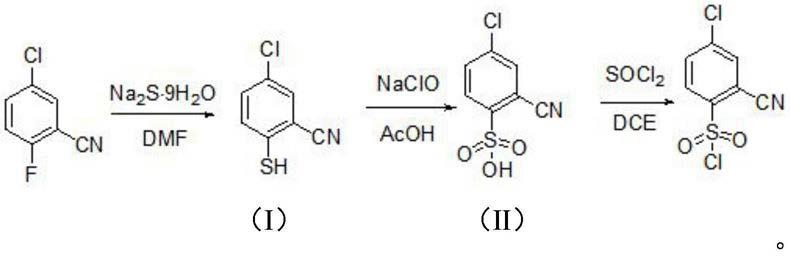
一方面,本发明涉及一种4
‑
氯
‑2‑
氰基苯磺酰氯的合成方法,该方法包括:在第一步骤中,将2
‑
氟
‑5‑
氯苯腈与九水硫化钠反应生成式(ⅰ)化合物;然后,在第二步骤中,将式(ⅰ)化合物氧化生成式(ⅱ)化合物;最后,在第三步骤中,将式(ⅱ)化合物与二氯亚砜反应生成4
‑
氯
‑2‑
氰基苯磺酰氯,
[0007]
式(ⅰ)化合物为:
[0008][0009]
式(ⅱ)化合物为:
[0010][0011]
另一方面,本发明还提供了4
‑
氯
‑2‑
氰基苯磺酰氯的以下合成路线:
[0012][0013]
另一方面,本发明提供了合成式(ⅰ)化合物的方法,具体包括:惰性气体保护下,将2
‑
氟
‑5‑
氯苯腈与九水硫化钠溶于dmf后控温50~55℃反应6h得到反应液1,将反应液1经冰水处理、碱化、二氯甲烷萃取除杂、酸化、过滤、二氯甲烷溶解、水洗、干燥、浓缩、正庚烷打浆、过滤、干燥得到式(ⅰ)化合物。
[0014]
进一步地,以摩尔质量计,2
‑
氟
‑5‑
氯苯腈与九水硫化钠的摩尔比为1:1.2;以质量份计,2
‑
氟
‑5‑
氯苯腈与dmf质量比为1:9.4。
[0015]
另一方面,本发明提供了合成式(ⅱ)化合物的方法,具体包括:惰性气体保护下,将式(ⅰ)化合物与乙酸混合后,滴加氧化剂,控温20~30℃反应4h得到反应液2,将反应液2经亚硫酸钠破坏后,浓缩至糊状,甲醇打浆、过滤、浓缩、降温过滤得到式(ⅱ)化合物;优选地,氧化剂选用次氯酸钠。
[0016]
进一步地,以摩尔质量计,式(ⅰ)化合物与次氯酸钠摩尔比为1:3;以质量份计,式(ⅰ)化合物与乙酸的质量比为1:10.5。
[0017]
进一步地,本发明提供了式(ⅱ)化合物合成4
‑
氯
‑2‑
氰基苯磺酰氯的方法,具体包括:惰性气体保护下,以二氯乙烷为液体环境,将式(ⅱ)化合物与氯代试剂混合,控温62~72℃反应13h制得反应液3,将反应液3经冰水处理、萃取、有机相水洗、干燥、浓缩、重结晶、过滤、干燥得到4
‑
氯
‑2‑
氰基苯磺酰氯;优选地,氯代试剂选用二氯亚砜。
[0018]
进一步地,以摩尔质量计,所述式(ⅱ)化合物与氯代试剂的摩尔比为1:6.01;以质量份计,所述式(ⅱ)化合物与二氯乙烷的质量比为1:12.4。
[0019]
本发明与现有技术相比具有以下有益效果或者优点:
[0020]
(1)本发明提供的4
‑
氯
‑2‑
氰基苯磺酰氯合成路线,避免了重氮化反应的发生,提高了生产的安全性;
[0021]
(2)本发明提供的4
‑
氯
‑2‑
氰基苯磺酰氯合成路线,所用原料简单,低毒,仅只会产生少量的含氮及含醋酸的废水;
[0022]
(3)本发明提供的4
‑
氯
‑2‑
氰基苯磺酰氯合成路线,仅为三步反应,且每步反应的
反应时间及后处理时间均较短,后处理简单,产品的制造成本较低。
附图说明
[0023]
图1为实施例1制得的4
‑
氯
‑2‑
氰基苯磺酰氯的lc图谱;
[0024]
图2为实施例1制得的4
‑
氯
‑2‑
氰基苯磺酰氯的核磁氢谱图。
具体实施方式
[0025]
下面,结合实施例对本发明的技术方案进行说明,但是,本发明并不限于下述的实施例。
[0026]
实施例1
[0027]
本实施例提供了一种4
‑
氯
‑2‑
氰基苯磺酰氯的合成方法、详细过程和表征分析,包括其合成前体式(ⅰ)化合物与式(ⅱ)化合物的合成方法和详细过程,具体如下:
[0028]
(1)式(ⅰ)化合物的合成
[0029][0030]
氮气保护下,向装有机械搅拌、温度计的2000ml三口瓶中依次加入dmf 1500ml,2
‑
氟
‑5‑
氯苯腈150.0g,搅拌溶解澄清后,加入na2s
·
9h2o 277.91g,加完料后缓慢加热升温至50℃~55℃保温反应,当2
‑
氟
‑5‑
氯苯腈gc<1%时,停止反应。
[0031]
反应完毕后,在氮气保护下,将反应液自然降温至20℃~25℃,倒入搅拌的1200ml冰水中,搅拌5min,冰浴条件下控温在10℃~20℃,开始滴加1mol/l氢氧化钠水溶液1050ml,搅拌15min后,用二氯甲烷1200ml
×
2次萃取除去反应产生的杂质,分液,收集水相。向水相中缓慢滴加盐酸450ml,过程控温10℃~20℃,调至体系ph=1~2后,搅拌20min,过滤收集滤固。将收集的滤固加入到二氯甲烷750ml中,搅拌溶解清亮,水洗至ph=6~7,搅拌20min,静置10min,分液,收集有机相。向有机相中加入无水硫酸镁搅拌干燥,过滤、滤液浓缩至有机相剩余二氯甲烷150ml,加入正庚烷450ml,继续浓缩至剩余约200ml正庚烷后,放入冰箱(
‑
20℃~
‑
15℃)冷冻8h,过滤,至式(ⅰ)化合物与式(ⅰ)化合物偶联产物总含量gc>70%。将过滤后的产物进行烘料(30℃~35℃,
‑
0.09mpa~
‑
0.07mpa,9h)至溶剂残留<0.5%。
[0032]
经检测,式(ⅰ)化合物含量+式(ⅰ)化合物偶联产物含量gc=19.3735%+54.4325%=73.8060%,烘料溶剂残留=0.1341%,得棕黄色固体126.3g,收率:77.22%(以2
‑
氟
‑5‑
氯苯腈计)。
[0033]
(2)式(ⅱ)化合物的合成
[0034][0035]
向装有机械搅拌、温度计、恒压滴液漏斗的2000ml三口瓶中加入乙酸500ml,式(ⅰ)化合物+式(ⅰ)化合物偶联产物50.00g,搅拌10min后,用冰水浴控温在20℃~30℃开始滴加次氯酸钠溶液(实测次氯酸钠含量为9.2%)716.1g,滴加完毕后,在20℃~30℃条件下搅拌反应,当原料式(ⅰ)化合物+式(ⅰ)化合物偶联产物总含量lc<1%时,停止反应。
[0036]
反应完毕后,加入无水亚硫酸钠5.0g淬灭反应,浓缩淬灭后的反应液至糊状,使剩余液体50ml,加入甲醇500ml,继续进行浓缩至有大量固体析出,液体剩余25ml后,停止浓缩。用甲醇750ml,40℃~45℃条件下打浆,搅拌30min,静置10min,过滤,收集滤液,将滤固再次用甲醇750ml,40℃~45℃条件下打浆,搅拌30min,静置10min,过滤。合并收集两次滤液,浓缩滤液至有大量白色固体析出,剩余甲醇10ml后,再加入乙酸乙酯200ml,浓缩至剩余液体100ml后,停止浓缩。将剩余液在室温下搅拌20min,静置10min,过滤,至式(ⅱ)化合物lc>96%,烘料(35℃~40℃,
‑
0.09mpa~
‑
0.07mpa,约8h)至溶剂残留<0.5%。
[0037]
经检测,实测溶剂残留=0.0208%,得白色固体70.5g,以式(ⅰ)化合物计,收率为100%。
[0038]
(3)4
‑
氯
‑2‑
氰基苯磺酰氯的合成
[0039][0040]
氮气保护下,向装有机械搅拌、温度计、恒压滴液漏斗的2000ml三口瓶中加入二氯乙烷600ml,式(ⅱ)化合物60.00g,搅拌10min后,开始滴加氯化亚砜197.01g,氯化亚砜加完后,搅拌5min后,滴加dmf 12ml,滴加完毕后,缓慢加热升温至回流62℃~72℃保温反应,当式(ⅱ)化合物lc<2%时,停止反应。
[0041]
反应完毕后,自然降温至20℃~30℃,将反应液倒入冰水500ml中,搅拌40min至清亮,分液,得有机相,水相再用二氯甲烷600ml
×
1次萃取,将所得有机相合并水洗,每次搅拌5min,静置10min,分液,后有机相中加入无水硫酸镁12.00g搅拌干燥1h后,过滤,有机相浓缩至无溶剂馏出,得黄色油状粗品45.5g。
[0042]
向上述黄色油状物中加入正己烷120ml,在冰浴条件(0℃~10℃)下搅拌重结晶,搅拌1h后,放入冰箱冷冻(
‑
15℃~
‑
20℃)4h,过滤,重结晶至4
‑
氯
‑2‑
氰基苯磺酰氯lc>98%时,烘料(30℃~35℃,
‑
0.09mpa~
‑
0.07mpa,约8h)至溶剂残留≤5000ppm,得类白色固体即为所述4
‑
氯
‑2‑
氰基苯磺酰氯。
[0043]
经检测,4
‑
氯
‑2‑
氰基苯磺酰氯lc=99.2256%,溶剂残留=767ppm,得类白色固体33.0g,以式(ⅱ)化合物计,收率为55.72%。
[0044]
(4)步骤(3)所得4
‑
氯
‑2‑
氰基苯磺酰氯的表征分析
[0045]
图1是步骤(3)制得的4
‑
氯
‑2‑
氰基苯磺酰氯的lc图谱。由图1可得,得到的目标产物lc纯度99.2256%,产品lc含量高。图2是步骤(3)制得的4
‑
氯
‑2‑
氰基苯磺酰氯的核磁氢谱图,由图2可得:δ7.71ppm~7.73ppm(s,1h,ch),7.73ppm~7.84ppm(d,1h,ch),7.84ppm~7.95ppm(d,h,ch),符合目标产物的核磁氢谱图。
[0046]
由此可得,本发明以2
‑
氟
‑5‑
氯苯腈为原料经三步反应合成4
‑
氯
‑2‑
氰基苯磺酰氯,所用的原材料简单,低毒,仅只会产生少量的含氮及含醋酸的废水;本发明常规反应设备即可满足工业化生产的要求,工厂也不需要特殊的资质,得到的产物纯度高达99%以上,且品质稳定,重复性好。由于该合成路线所用的原料简单,价格低廉,且合成总收率能到43.03%,故单位产品的原材料成本较低;由于合成路线仅为三步反应,且每步反应的反应时间及后处理时间均较短,后处理简单,故单位产品的制造成本较低;综上,该方法适合工业化生产。
[0047]
如上所述,即可较好地实现本发明,上述的实施例仅仅是对本发明的优选实施方式进行描述,并非对本发明的范围进行限定,在不脱离本发明设计精神的前提下,本领域普通技术人员对本发明的技术方案做出的各种改变和改进,均应落入本发明确定的保护范围内。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1