基于单量子点的多引物生成滚环扩增纳米传感器及单核苷酸多态性检测方法
1.本发明属于生物检测和分子生物学技术领域,具体涉及一种基于单量子点的多引物生成滚环扩增纳米传感器及单核苷酸多态性检测方法。
背景技术:
2.公开该背景技术部分的信息仅仅旨在增加对本发明的总体背景的理解,而不必然被视为承认或以任何形式暗示该信息构成已经成为本领域一般技术人员所公知的现有技术。
3.单核苷酸多态性(snp)是广泛研究的人类遗传变异。单核苷酸多态性发生在基因组序列的特定位置,主要包括单核苷酸替换、插入和缺失。大多数snp是沉默的,不会改变基因的功能和表达。单核苷酸多态性与癌症、哮喘、神经性疾病、镰状细胞贫血和阿尔茨海默病密切相关,甚至导致患者的药物反应变化。单核苷酸多态性可以作为识别许多遗传疾病的生物标志物。因此,灵敏和准确地测定snps对于疾病的早期临床诊断、风险评估和预后监测至关重要。
4.传统的单核苷酸多态性检测方法包括直接测序、单链构象多态性(sscp)分析、高分辨率熔解曲线分析(hrma)、变性高效液相色谱(dhplc)和基于分子信标的方法。直接测序尽管在发现新的单核苷酸多态性方面很有价值,但它仍然是一个昂贵且耗时的方法;传统的二代测序仅限于相对较高比例的突变型到野生型dna(>1%),不适合于血液中的ctdna分析。sscp分析采用半自动电泳系统和凝胶/毛细管电泳,能检测任何特定位点的dna序列改变,但它仅适用于定性分析。hrma方法可以根据突变靶点和野生型靶点之间的熔解曲线和熔解温度的差异来检测snps,但它涉及高严格性条件和精确的温度控制。dhplc能够直接检测snps,具有快速可靠的特点,但它涉及昂贵的仪器,难以区分不同的突变类型。基于分子信标的方法尽管简单,但由于不完全猝灭,背景信号高,灵敏度有限。近年来,一系列的dna扩增技术已经用于检测单核苷酸多态性,包括聚合酶链反应(pcr)、数字pcr(dpcr)和自主等温聚合。然而,由于昂贵的设备和外源性引物的介入,这些方法很难并行化,并且容易产生dna样本污染,从而导致引物二聚化和非特异性扩增。近期,现有技术中衍生出基于滚环扩增的荧光检测法,这种方法一般涉及到荧光基团和猝灭剂修饰的探针或sybr gold染料,不可避免的产生背景信号。而且,荧光方法与单分子方法相比,灵敏度较低,样品消耗量大。因此,迫切需要一种简便、灵敏的单核苷酸多态性检测方法。
技术实现要素:
5.为了解决现有技术的不足,本发明提供一种基于单量子点(qd)的多引物生成滚环扩增纳米传感器,该传感器集成了多引物生成的滚环扩增反应(mpg
‑
rca),用于灵敏检测snp。该传感器仅涉及一个挂锁探针,存在突变靶点时,线性挂锁探针环化以启动rca,在核酸内切酶的帮助下产生三种类型的短单链dna(ssdnas),由此产生的ssdna可以作为引物诱
导循环的mpg
‑
rca反应,导致指数扩增并产生大量连接探针。连接探针、cy5标记的报告探针、生物素化捕获探针可在量子点表面上组装形成三明治夹心结构结构,导致从量子点供体到cy5受体的有效荧光共振能量转移(fret),进而实现灵敏检测snp。
6.具体包括如下技术方案:
7.本发明第一方面提供一种基于单量子点的多引物生成滚环扩增纳米传感器,包括量子点和挂锁探针,所述挂锁探针由两个突变目标互补序列、三个核酸内切酶识别位点和两个启动mpg
‑
rca的区域组成。
8.本发明第二方面提供一种单核苷酸多态性检测方法,具体包括如下步骤:
9.(1)点突变序列驱动线性挂锁探针的连接,生成环状探针;
10.(2)通过mpg
‑
rca反应引发连接探针大量积累;
11.(3)605qd
‑
dna
‑
cy5纳米结构的形成;
12.(4)单分子成像。
13.本发明的一个或多个实施方式至少具有以下有益效果:
14.该纳米传感器不需要复杂的探针设计或外源性引物,具有扩增效率高、背景信号零、特异性好、灵敏度高、无需任何洗涤/分离步骤即可进行均相分析等显著优点。它可以检测到8个数量级的大动态范围的snp,检测限为5.41
×
10
‑
20
mol/l。此外,这种纳米传感器可以从混合物中准确地区分低至0.001%的突变水平,这是以前报道的方法无法实现的。此外,它可以区分正常细胞和癌细胞,甚至在单细胞水平上量化snp。
附图说明
15.构成本发明的一部分的说明书附图用来提供对本发明的进一步理解,本发明的示意性实施例及其说明用于解释本发明,并不构成对本发明的不当限定。
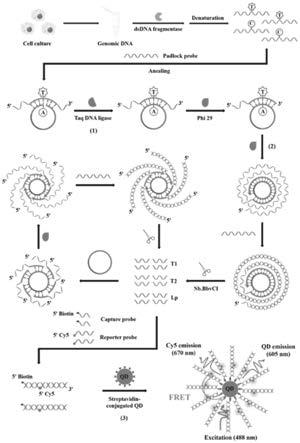
16.图1为基于单量子点的多引物生成滚环扩增纳米传感器用于单核苷酸多态性检测的原理图。
17.图2为可行性分析系列图,其中:(a)以sybrgold为指示剂的突变靶标驱动挂锁探针连接的page分析。lanem,dnamarker;泳道1,野生型靶标;泳道2,突变型靶标。(b)突变靶标启动mpg
‑
rca产物的琼脂糖凝胶电泳分析。lane m,2000bp dnamarker;泳道1,突变型靶标;泳道2,野生型靶标。(c和d)rca产品(c)和mpg
‑
rca产品(d)的afm图像。(e和f)图像c和d中标记位置的高度分析。(g
‑
i)605qd
‑
dna
‑
cy5纳米结构的琼脂糖凝胶电泳分析。泳道1,突变型靶标;泳道2,野生型靶标;泳道3,605qd。605qd、cy5及其共定位的信号分别以绿色、红色和黄色显示。(j)cy5和605qd荧光发射光谱对野生型靶标和突变靶标的响应。插图显示了640
‑
720nm范围内的荧光发射光谱。本实验使用了100nm挂锁探针、10nm突变靶标和10nm野生靶标。
18.图3为605qd在没有突变靶标(对照)和存在突变靶标的情况下的荧光寿命曲线。605qd的浓度为5nm。
19.图4为分别响应野生型靶点(a
‑
c)和突变靶点(d
‑
f)的605qd(a和d)和cy5(b和e)的单分子图像。(c和f)表示cy5和605qd荧光信号的共定位。实验中使用了10nm野生型靶标、10nm突变靶标、100nm挂锁探针和5nm 605qd。比例尺为5μm。
20.图5为灵敏度检测相关图,其中:(a)cy5和605qd的荧光发射光谱随不同浓度突变
靶标的变化。插图显示了cy5在650
–
710nm范围内的荧光光谱。(b)cy5荧光强度与突变靶标浓度的对数呈线性相关。(c)突变靶标引发cy5计数的浓度依赖性增加。(d)在1
×
10
‑
19至1
×
10
‑
11m范围内,cy5计数与突变靶标浓度的对数呈线性相关。研究中使用了100nm挂锁探针和5nm 605qd。误差棒显示了三组独立实验的标准偏差。
21.图6为突变靶标、一个碱基错配dna(野生型靶)、两个碱基错配dna(错配
‑
1;错配
‑
2)和三碱基错配dna(错配
‑
3)产生的cy5数量。每种dna的浓度为10nm。误差棒显示了三组独立实验的标准偏差。
22.图7为测量值和实际加入突变水平之间的线性相关性。误差棒显示了三组独立实验的标准偏差。
23.图8为基因组dna突变检测的相关图,其中:(a)分别从panc
‑
1细胞、hct
‑
116细胞、a549细胞、hela细胞和hl
‑
7702细胞提取的基因组dna以及仅含裂解缓冲液的对照组产生的cy5数目的测量。细胞个数为10000个。(b)cy5计数与panc
‑
1细胞数的对数呈线性相关。误差棒显示了三组独立实验的标准偏差。
具体实施方式
24.应该指出,以下详细说明都是示例性的,旨在对本发明提供进一步的说明。除非另有指明,本文使用的所有技术和科学术语具有与本发明所属技术领域的普通技术人员通常理解的相同含义。
25.需要注意的是,这里所使用的术语仅是为了描述具体实施方式,而非意图限制根据本发明的示例性实施方式。如在这里所使用的,除非上下文另外明确指出,否则单数形式也意图包括复数形式,此外,还应当理解的是,当在本说明书中使用术语“包含”和/或“包括”时,其指明存在特征、步骤、操作、器件、组件和/或它们的组合。
26.正如背景技术所介绍的,现有技术中单核苷酸多态性检测方法存在操作复杂、条件严格、背景信号高、灵敏度低等诸多问题,目前迫切需要简便、灵敏的单核苷酸多态性检测方法。
27.为了解决如上的技术问题,本发明第一方面提供一种基于单量子点的多引物生成滚环扩增纳米传感器,包括量子点和挂锁探针,所述挂锁探针由两个突变目标互补序列、三个核酸内切酶识别位点和两个启动mpg
‑
rca的区域组成。
28.进一步的,所述纳米传感器还包括cy5标记的报告探针和生物素化捕获探针。
29.进一步的,所述量子点为605nm发射量子点。
30.本发明第二方面提供一种单核苷酸多态性检测方法,具体包括如下步骤:
31.(1)点突变序列驱动线性挂锁探针的连接,生成环状探针;
32.(2)通过mpg
‑
rca反应引发连接探针大量积累;
33.(3)605qd
‑
dna
‑
cy5纳米结构的形成;
34.(4)单分子成像。
35.上述检测方法具有如下几方面的优势:
36.1、与荧光检测相比,单分子检测有消耗样品量极少,信噪比高,灵敏度高,背景信号接近零的显著优点。
37.2、此发明仅需要使用一个挂锁探针,不涉及任何外源性引物,即可实现kras原癌
基因热点突变g12d的高灵敏和高特异性检测。
38.3、mpg
‑
rca循环扩增是一个具有高扩增效率的自动化系统,能达到指数扩增的效率,并且在等温条件下即可进行。
39.4、在单个qd表面组装多个三明治杂交体可获得单供体/多受体结构,大大提高fret效率,并有助于以高灵敏度均相检测snp。
40.进一步的,所述连接反应在含有1
×
tdt缓冲液、12u taq dna连接酶、1mm nad
+
、100nm挂锁探针和目标dna的反应溶液中进行;
[0041]1×
tdt缓冲液的组成为:10mm mg(ac)2、20mm tris ac、50mm kac,ph为7.9。
[0042]
进一步的,所述连接反应发生的条件为:90
‑
95℃下孵育5
‑
10min,然后在40
‑
45℃下孵育50
‑
60min。
[0043]
进一步的,步骤(2)中,向环状探针中添加0.5μl 10
×
tdt缓冲液、3u nb.bbvci、1u phi29 dna聚合酶和50μm dntps,并在35℃下培养90分钟以进行mpg
‑
rca反应,然后在75
‑
85℃下酶失活15
‑
25分钟,进行连接探针的大量积累。
[0044]
其中,通过添加phi29 dna聚合酶对目标dna的3'端悬垂核苷酸进行修剪,以形成修剪后的目标dna/环状探针杂交体,同时,在phi29 dna聚合酶存在下启动rca反应,产生包含与环状探针互补的重复单元的长单链dna(ssdna);phi29 dna聚合酶能够将不同长度的含有突变靶标的片段转化为rca引物,有助于直接在各种基因组dna中检测突变靶标。
[0045]
生成的长单链dna产物与游离的环状探针杂交,形成串联的dna双链,其中包含多个内切酶nb.bbvci识别位点。nb.bbvci切割dna双链的特定位置以产生大量的三种类型的短单链dna产物,即触发探针1、触发探针2和连接探针,这些短ssdna产物可作为新引物与环状探针配对(复合物ii),启动多个mpg
‑
rca反应循环,其可以指数方式放大单个rca循环,以产生大量包含连接探针的短ssdna产物。
[0046]
进一步的,步骤(3)中,将mpg
‑
rca反应产物与10μm生物素化的捕获探针和10μm cy5修饰的报告探针一起于90
‑
95℃下孵育5
‑
8min,然后冷却至室温以获得夹心杂交结构。
[0047]
mpg
‑
rca反应产物、10μm生物素化的捕获探针、10μm cy5修饰的报告探针的体积比为30:1.44:1.44;
[0048]
进一步的,将夹心杂交结构与包被有链霉亲和素的605qd在室温下孵育10
‑
20分钟,获得605qd
‑
dna
‑
cy5纳米结构;
[0049]
进一步的,全内反射荧光(tirf)显微镜用于单分子成像。
[0050]
为了使得本领域技术人员能够更加清楚地了解本发明的技术方案,以下将结合具体的实施例详细说明本发明的技术方案。
[0051]
1、实验方案
[0052]
(1)细胞培养与基因组dna提取:
[0053]
hct
‑
116细胞在含有1%青霉素
‑
链霉素和10%胎牛血清(美国gibco)的mccoy 5a培养基中培养。在包含1%青霉素
‑
链霉素和10%胎牛血清的dulbecco改良eagle培养基(dmem)中培养panc
‑
1细胞、a549细胞、hela细胞和hl
‑
7702细胞。所有细胞均在37℃、含5%co2的潮湿培养箱中培养。使用qiaamp dna微试剂盒提取基因组dna,并使用双链dna片段化酶对长dna链进行片段化。使用nanodrop 2000分光光度计对基因组dna浓度进行定量。
[0054]
(2)连接反应和mpg
‑
rca反应:
[0055]
连接反应在含有1
×
tdt缓冲液(10mm mg(ac)2、20mm tris ac、50mm kac、ph 7.9)、12u taq dna连接酶、1mm nad
+
、100nm挂锁探针和目标dna(或从细胞中提取的基因组dna)的反应溶液(10μl)中于95℃下孵育5min,然后在45℃下孵育60分钟。连接后,向混合物中添加0.5μl 10
×
tdt缓冲液、3u nb.bbvci、1u phi29 dna聚合酶和50μm dntps,并在35℃下培养90分钟以进行mpg
‑
rca反应,然后在80℃下酶失活20分钟。
[0056]
(3)605qd
‑
dna
‑
cy5纳米结构的构建:
[0057]
将mpg
‑
rca反应产物(30μl)与10μm生物素化的捕获探针(1.44μl)和10μm cy5修饰的报告探针(1.44μl)在缓冲溶液(3mm mgcl2,10mm(nh4)2so4,100mm tris hcl,ph 8.0)中于95℃下孵育5min,然后冷却至室温以获得夹心杂交结构。将夹心杂交体与0.2μm包被有链霉亲和素的605qd(2μl)在室温下孵育15分钟,获得605qd
‑
dna
‑
cy5纳米结构。
[0058]
(4)凝胶电泳和原子力显微镜测量:
[0059]
为了验证连接反应的特异性,用核酸外切酶i和iii消化连接产物,然后用12%聚丙烯酰胺凝胶电泳(page)于1
×
tbe缓冲液(0.2mm edta,9mm硼酸,9mm tris
‑
hcl,ph 7.9)中在110v恒定电压下室温下以sybr gold为指示剂电泳45min。为了验证扩增反应,扩增产物在110v恒定电压下,1
×
tae缓冲液(2mm edta和40mm tris
‑
hac)中通过1%琼脂糖凝胶电泳60min。为了验证605qd
‑
dna
‑
cy5纳米结构的构建,反应产物在110v恒定电压下在1
×
tae缓冲液中通过2%琼脂糖凝胶电泳50分钟进行分析。凝胶成像使用bio
‑
rad chemidoc成像系统。
[0060]
我们使用原子力显微镜(afm)对突变突变体启动的mpg
‑
rca产物进行了表征。dna样品固定在原子级平坦的云母表面上。将mpg
‑
rca产物稀释至1ng/μl,并在室温下沉积在3
‑
氨丙基三乙氧基硅烷(aptes)
‑
修饰的云母上5分钟,然后用去离子水冲洗并用氮气干燥。采用veeco di纳米镜多模v系统进行afm测量。
[0061]
(5)荧光测量:
[0062]
荧光发射光谱由fls1000荧光仪测量,激发波长为488nm,激发和发射狭缝均为2.5nm。分别使用605nm处的605qd荧光强度和670nm处的cy5荧光强度进行数据分析。采用时间相关单光子计数法(tcspc)测量了605qds的荧光寿命。
[0063]
(6)单分子检测与数据分析:
[0064]
全内反射荧光(tirf)显微镜用于单分子成像。在200倍稀释后,将10μl反应产物滴在玻璃盖玻片上,以488nm蓝色激光作为激发源进行成像。使用100
×
油浸物镜收集发射的光子,并使用二向色镜将荧光信号分为cy5通道和605qd通道。曝光时间为500ms,使用emccd摄像机获取荧光图像。使用image j软件对600
×
600像素区域内的cy5分子进行计数,并使用十帧计数的平均值进行数据分析。
[0065]
2、snp分析的原理介绍
[0066]
我们选择kras癌基因热点突变g12d(kras外显子2第12密码子突变)作为本研究的靶点。snp分析的原理如图1所示。mpg
‑
rca系统由一个突变靶点和一个挂锁探针组成。所设计的突变体和野生型靶标在5'端和3'端分别有6个核苷酸尾以模拟实际样品中的dna片段。挂锁探针由位于5'和3'端的两段突变性目标互补序列、nb.bbvci核酸内切酶的三个识别位点和mpg
‑
rca启动的两个特殊区域组成。该方法包括三个步骤:(1)点突变序列驱动线性挂锁探针的连接,(2)mpg
‑
rca反应引发的连接探针大量积累,(3)605qd
‑
dna
‑
cy5纳米结构的
形成和单分子成像。在第一步中,kras突变序列与线性挂锁探针的两端杂交,使5'和3'端接近,在taq dna连接酶的作用下连接,生成环状探针。突变靶标的3'端悬垂核苷酸可通过phi29 dna聚合酶进行修剪,以形成修剪后的突变靶标/环状探针杂交体(复合物i)。复合物i可以引发以下级联反应。首先,在phi29 dna聚合酶存在下启动rca反应,产生包含与环状探针互补的重复单元的长单链dna(ssdna)。其次,生成的长单链dna产物与游离的环状探针杂交,形成串联的dna双链,其中包含多个内切酶nb.bbvci识别位点。第三,nb.bbvci切割dna双链的特定位置以产生大量的三种类型的短单链dna产物,即触发探针1(t1)、触发探针2(t2)和连接探针(lp)。第四,这些短ssdna产物可作为新引物与环状探针配对(复合物ii),启动多个mpg
‑
rca反应循环(即t1、t2和lp启动的mpg
‑
rca),其可以指数方式放大单个rca循环,以产生大量包含连接探针的短ssdna产物。随后,连接探针可与cy5标记的报告探针和生物素化的捕获探针杂交形成三明治结构。通过链霉亲和素
‑
生物素特异性相互作用在605qd表面组装三明治结构,获得605qd
‑
dna
‑
cy5纳米结构,从而实现从605qd到cy5的高效fret,通过单分子检测可以准确地测定突变水平。相反,当存在野生型靶标时,由于连接部位存在腺嘌呤和胞嘧啶错配,连接反应无法发生,605qd
‑
dna
‑
cy5纳米结构的形成以及605qd到cy5的fret都无法发生,就不会产生信号。
[0067]
3、可行性分析
[0068]
本方法提出的纳米传感器依赖于突变靶标介导的挂锁探针特异性连接、mpg
‑
rca的高效率扩增、605qd
‑
dna
‑
cy5纳米结构的构建以及605qd和cy5之间的高效fret。为了研究突变靶标是否能引发挂锁探针的连接,我们采用聚丙烯酰胺凝胶电泳(page)来检测连接产物。当存在野生型靶点时,腺嘌呤(a)
‑
胞嘧啶(c)的单一错配不能引发连接反应。如图2所示,经核酸外切酶处理后,未检测到条带(图2a,泳道1),因为野生型靶标和带有3'
‑
oh末端的挂锁探针全都被核酸外切酶i和核酸外切酶iii消化。当存在突变靶标时,挂锁探针连接成环形。通过核酸外切酶处理后,能观察到一条环状挂锁探针带(图2a,泳道2),因为核酸外切酶i和iii可以消化线性寡核苷酸(例如突变靶标),而不是环状挂锁探针。我们使用琼脂糖凝胶电泳来测量mpg
‑
rca产物(图2b)。当存在野生型靶标时,检测到单一条带(图2b,泳道2),表明未发生mpg
‑
rca反应。相反,当存在突变靶点时检测到扩增产物的条带(图2b,泳道1),表明发生突变靶点引发的mpg
‑
rca反应。使用原子力显微镜(afm)对mpg
‑
rca产物进行了进一步表征。afm图像显示,由于rca产生的单链dna的灵活性,存在长度为几微米的长线性和分支结构(图2c)。与rca产物的长单链dna产物相比(图2c),观察到mpg
‑
rca的产物是大量短单链dna产物(图2d),每个单链dna的高度测量为1.2nm(图2e和2f),表明是单层的单链dna。这些结果表明:(1)突变靶标诱导的挂锁探针连接的可行性;(2)点突变驱动的连接反应具有良好的特异性;(3)突变靶标启动的mpg
‑
rca具有较高的扩增效率。
[0069]
琼脂糖凝胶电泳证实了605qd
‑
dna
‑
cy5纳米结构的成功构建。单独的包被有链霉亲和素的605qd不带电,产生具有低电泳迁移率的绿色条带(图2g,泳道3)。在突变靶标存在的情况下,连接探针与cy5标记的报告探针和生物素化的捕获探针杂交获得605qd
‑
dna
‑
cy5纳米结构,黄色条带(图2i,泳道1)表示605qd(图2g,泳道1)和cy5(图2h,泳道1)的共定位。当存在野生型靶标时,生物素化的捕获探针在605qd表面上的组装获得605qd
‑
capture probe纳米结构,仅观察到605qd的绿色条带(图2g
‑
2i,泳道2)。此外,605qd
‑
dna
‑
cy5纳米结构(图2g和2i,泳道1)的条带迁移速度比605qd
‑
capture probe纳米结构(图2g和2i,泳道2)
慢,表明605qd
‑
dna
‑
cy5纳米结构组装成功。
[0070]
为了研究突变靶标能否引发从605qd到cy5的有效fret,我们测量了野生型和突变型的荧光发射光谱(图2j)。当存在野生型靶标时,仅检测到605qd荧光信号,未观察到cy5荧光信号(图2j)。当存在突变靶点时,605qd荧光信号降低,同时出现cy5荧光信号(图2j),表明突变靶点诱导产生605qd和cy5之间的有效fret。
[0071]
4、量子点纳米传感器的fret效率计算
[0072]
在具有一个供体和多个受体的605qd
‑
dna
‑
cy5纳米结构中,理论fret效率(e)可以根据方程(1)计算
[0073][0074]
其中r是供体和受体平均距离,n是每个供体上的受体数,r0是距离。链霉亲和素包被的605qd的半径r1=7.5nm,根据dsdna中相邻碱基之间的0.34nm,计算605qd表面与cy5分子之间的距离r2=5.44nm。r计算为12.94nm(r1+r2),在有效fret范围内(2r0=15.4nm),表明可以发生从605qd到cy5的有效fret。值得注意的是,单个605qd可连接36个生物素化cy5标记的三明治探针,提高了等式(1)的605qd
‑
dna
‑
cy5纳米结构中的fret效率。理论fret效率计算为61.5%。此外,在优化后的条件下,可通过使用荧光发射光谱对fret效率进行实验测量,根据方程(2)
[0075][0076]
其中fd和fda分别是在没有和存在突变靶标的情况下的605qd荧光强度。测量的e值为64.0%,大于理论fret效率(61.5%)。这可以用dna的弯曲灵活性来解释,dna的灵活性可能导致cy5受体与605qd供体更接近。此外,可通过基于荧光寿命的实验测量fret效率,根据方程(3)
[0077][0078]
式中,τ
d
和τ
da
分别是在没有和存在突变靶标情况下的605qd荧光寿命。如图3所示,突变靶标的存在导致605qd的平均寿命从31.4ns(对照组,图3)降低到11.1ns(图3)。fret效率为64.6%,与荧光发射光谱测量结果(64.0%)一致。
[0079]
5、单核苷酸多态性的单分子成像
[0080]
我们使用tirf显微镜对snp进行单分子成像。如图4所示,野生型靶标仅产生605qd荧光信号(图4a)而没有cy5荧光信号(图4b),表明没有cy5分子组装到605qd表面,605qd和cy5之间没有发生fret。相反,当突变靶标存在时,由于605qd到cy5发生有效fret,605qd(图4d)和cy5(图4e)荧光信号同时出现,605qd和cy5荧光信号具有明显的共定位(图4f)。此外,野生型靶标显示出绝对零背景信号(图4b),表明该纳米传感器对突变靶标具有良好的特异性。
[0081]
6、检测灵敏度
[0082]
我们测量了不同浓度突变靶标产生的cy5荧光强度。如图5a所示,突变靶标浓度升高时,605qd荧光强度逐渐降低,而cy5荧光强度同时增加,表明突变靶标成功引发605qd到cy5的有效fret。此外,在1
×
10
‑
18
m至1
×
10
‑
11
m范围内,cy5荧光强度与突变靶标浓度的对数之间呈现良好的线性相关性(图5b),回归方程为f=62561.31+3381.12log
10 c(r2=
0.9967),其中c代表突变靶标浓度(m),f代表cy5荧光强度。通过计算对照组的平均响应信号加上三倍标准偏差,确定检测限为5.89
×
10
‑
19
m。此外,我们还监测了不同浓度突变靶标产生的cy5数量。随着突变靶标浓度增加,cy5荧光分子数也不断增加(图5c)。对数坐标中,在1
×
10
‑
19
m到1
×
10
‑
11
m的范围内,cy5计数和突变靶标浓度之间呈现线性相关性(图5d),回归方程为n=738.65+37.37log
10 c(r2=0.9957),其中c是突变靶标浓度(m),n是测得的cy5分子数。检测限为5.41
×
10
‑
20
m。值得注意的是,该纳米传感器的灵敏度比基于aunp的比色纳米传感器(2
×
10
‑9m)高10个数量级,比基于分子信标的荧光生物传感器(2
×
10
‑
11
m)高8个数量级,比基于aunp的荧光纳米传感器(4
×
10
‑
12
m)高7个数量级,比基于靶循环连接的荧光生物传感器(6
×
10
‑
13
m)高7个数量级,比基于dnazyme的化学发光生物传感器(1
×
10
‑
16
m)高3个数量级。此外,单分子检测(图5d)的灵敏度比荧光光谱法(图5b)的灵敏度高10.89倍。灵敏度的提高可归因于(1)taq dna连接酶介导的连接反应的高特异性,(2)基于单个qd的fret的绝对零背景,(3)mpg
‑
rca的高扩增效率,(4)每个qd供体的多个cy5受体导致的fret效率的提高,以及(5)单分子检测的高信噪比。
[0083]
7、检测选择性
[0084]
我们使用一个碱基错配dna(即野生型靶标)、两个碱基错配dna(即错配
‑
1和错配
‑
2)和三个碱基错配dna(即错配
‑
3)作为干扰物来评估该纳米传感器的选择性(图6)。只有突变靶标可以产生高的cy5信号(图6),但野生型靶标(图6)、错配
‑
1(图6)、错配
‑
2(图6)和错配
‑
3(图6)均不能产生显著的cy5信号,这表明该纳米传感器对突变靶标的检测具有良好的特异性。
[0085]
8、稀有dna等位基因频率的检测
[0086]
snps通常只占样本总dna中的一小部分,从高背景的野生型靶点区分低含量的突变靶点至关重要。为了研究这种纳米传感器检测稀有dna等位基因频率的可行性,我们将突变靶点和野生型靶点以不同的比例混合,分别含有0.001%、0.01%、0.1%、1%、10%和100%的突变靶点。根据等式(4)计算测得突变水平:
[0087]
突变水平(%)=m/(m+w)
×
100%
ꢀꢀꢀ
(4)
[0088]
其中,m是测量出的突变靶标数量,w是野生型靶标的数量。当加入的突变水平从0.001%增加到100%时,测得的突变水平呈现线性升高(图7),回归方程为y=1.02x
–
0.16(r2=0.9976),其中x和y分别为加入和测得的突变水平。此纳米传感器可以很好地区分低至0.001%的等位基因频率,这是以前报道的方法无法实现的,如错配延伸和基于aunp的荧光纳米传感器(5%)、基于分子信标的实时pcr方法(2%)和基于靶循环连接的荧光分析(0.75%)。
[0089]
9、基因组dna突变的检测
[0090]
我们进一步检测了各种细胞系的dna突变水平,包括人胰腺癌细胞系(panc
‑
1细胞)、人结直肠癌细胞系(hct
‑
116细胞)、人肝细胞系(hl
‑
7702细胞)、人宫颈癌细胞系(hela细胞)和人肺腺癌细胞系(a549细胞)。由于基因组dna片段化过程中dna断裂位点的不确定性,经双链dna片段化酶处理后可能产生不同长度含有突变靶点的dna片段。值得注意的是,phi29 dna聚合酶有校对功能。在phi29 dna聚合酶的帮助下,含有突变靶点的dna片段的所有悬垂3'端可有效转化为引物,其随后可启动mpg
‑
rca反应并最终诱导605qd
‑
dna
‑
cy5纳米结构的形成。如图8a所示,包括panc
‑
1细胞(图8a)、hct
‑
116细胞(图8a)、a549细胞(图8a)和
hela细胞(图8a)的癌细胞系产生不同的cy5信号。相比之下,正常细胞系hl
‑
7702(图8a)产生低的cy5信号,与仅含有裂解缓冲液的对照组相同(图8a),表明该纳米传感器能够区分正常细胞和癌细胞。我们进一步定量检测panc
‑
1细胞中的kras g12d突变水平(图8b)。在1
‑
10000个细胞范围内,cy5计数与panc
‑
1细胞数对数之间存在良好的线性相关性,回归方程为n=51.51+29.51log
10 x,相关系数为0.9961,其中x为panc
‑
1细胞数,n为测量的cy5计数。值得注意的是,所提出的纳米传感器可以在单细胞水平检测snp,在癌症的早期临床诊断中具有巨大的前景。
[0091]
实施例中涉及的核苷酸序列如表1所示:
[0092]
表1
[0093][0094]
在kras野生型靶标和突变靶标中,下划线的“c”和“t”分别表示snp位点。在错配的dna中,下划线表示错配的碱基。在挂锁探针中,“p”表示5'末端的磷酸基团(po4)修饰,粗体表示与突变靶标的杂交区域。
[0095]
以上所述仅为本发明的优选实施例而已,并不用于限制本发明,对于本领域的技术人员来说,本发明可以有各种更改和变化。凡在本发明的精神和原则之内,所作的任何修改、等同替换、改进等,均应包含在本发明的保护范围之内。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1