苯并异硒唑酮类衍生物及其制备方法与应用
1.本发明属于有机化合物合成与医药应用技术领域,具体涉及一种含有苯并异硒唑酮衍生物及其制备方法与应用。
背景技术:
2.由新型冠状病毒(severe acute respiratory syndrome coronavirus 2,sars
‑
cov
‑
2)引起的covid
‑
19在全球范围内快速流行,对人类生命健康产生了严重威胁,目前仍缺少有效的治疗药物和具有100%抑制率的疫苗。
3.sars
‑
cov
‑
2的主蛋白酶(m
pro
)是一种半胱氨酸蛋白酶,在病毒复制和转录中起关键作用。此外,m
pro
在冠状病毒中是保守的,并且在不同冠状病毒中主蛋白酶的底物之间具有一些共同特征。因此,m
pro
是广谱抗冠状药物研发的重要靶点。
4.新型抗炎药物依布硒啉对sars
‑
cov
‑
2具有较好的抑制活性,但其临床效果仍需进一步评价。依布硒啉主要是通过硒原子与sars
‑
cov
‑
2 m
pro
的半胱氨酸的巯基形成共价键来抑制m
pro
活性。但体内的巯基众多,这种与巯基结合的抑制剂进入体内会大大降低其在体内的有效浓度,缺乏特异性,并容易产生脱靶效应。因此,如何增加依布硒啉与m
pro
活性位点的非共价键结合,以增加其选择性是研发m
pro
抑制剂所面临的难题。
5.本发明根据依布硒啉与sars
‑
cov
‑
2 m
pro
的结合特点与作用机制,通过合理药物设计、化学合成、生物活性评价发现了全新结构的苯并异硒唑酮衍生物,有望解决依布硒啉选择性差、易脱靶的问题。
6.
技术实现要素:
7.针对现有技术的不足,本发明提供了一种含有苯并异硒唑酮衍生物及其制备方法,本发明还提供上述化合物作为sars
‑
cov
‑
2 m
pro
抑制剂的活性筛选结果及其应用。
8.本发明的技术方案如下:
9.1.苯并异硒唑酮类衍生物
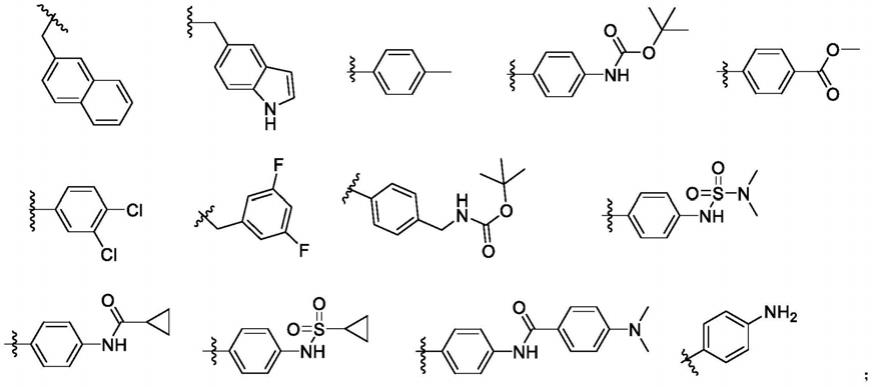
10.苯并异硒唑酮类衍生物,或其药学上可接受的盐、酯或前药,具有通式i所示的结构:
[0011][0012]
其中,
[0013]
r1为:取代苯基、取代苄基、萘亚甲基、喹啉亚甲基、吲哚亚甲基;所述的取代基选自甲基、叔丁氧羰基、n
‑
环丙甲酰胺基、n,n
‑
二甲基磺酰胺基、氨基、n
‑
环丙烷甲磺酰胺基、n,n
‑
二甲基苯甲酰胺基、甲酸乙酯基、卤素;
[0014]
r2为:硝基、氨基。
[0015]
根据本发明优选的,r1为下列取代基的任意一种:
[0016][0017]
r2为6
‑
硝基、6
‑
氨基。
[0018]
根据本发明进一步优选的,含有苯并异硒唑酮衍生物是下列化合物之一:
[0019]
表1.目标化合物苯并异硒唑酮类衍生物的结构
[0020]
[0021][0022]
[0023]
本发明中所述的“药学上可接受的盐”是指在可靠的医药评价范围内,化合物的盐类适于与人或较低等动物的组织相接触而无不适当的毒性、刺激及过敏反应等,具有相当合理的收益与风险比例,通常是水或油可溶的或可分散的,并可有效地用于其预期的用途。包括药学上可接受的酸加成盐和药学上可接受的碱加成盐,在这里是可做预期的用途并与式i、ii化合物的化学性质相容的。适宜的盐的列表参见s.m.birge等,j.pharm.sci.,1977,66,1
‑
19页。
[0024]
本发明中所述的“前药”是指药学上可接受的衍生物,以便这些衍生物所得的生物转换产物是如式i化合物所定义的活性药物。
[0025]
2.苯并异硒唑酮类衍生物的制备方法
[0026]
含有苯并异硒唑酮衍生物的制备方法,步骤如下:以邻碘苯甲酸i
‑
1为起始原料,二氯甲烷作为反应溶剂,通过酰胺缩合反应生成中间体(i
‑
2a~h,i
‑
11,i
‑
18);然后将中间体i
‑
2a~h溶解在适量的n,n
‑
二甲基苯甲酰胺中,经环合得到目标化合物(i
‑
3~i
‑
10);将中间体i
‑
11与i
‑
18溶解在适量二氯甲烷中,与相应的酸片段或酰氯片段反应得到关键中间体i
‑
12a~d与i
‑
21;接着,将经酰化反应得到的关键中间体溶解在适量的n,n
‑
二甲基苯甲酰胺中,经环合得到得到目标化合物(i
‑
13~i
‑
16,i
‑
22);将中间体i
‑
18溶解在适量的n,n
‑
二甲基苯甲酰胺中,经环合得到得到目标化合物(i
‑
19),然后在氯化亚锡的作用下发生还原得目标产物i
‑
20;
[0027]
合成路线如下:
[0028][0029]
试剂及条件:(i)r1‑
nh2,hatu,diea,二氯甲烷,室温;(ii)硒氰酸钾,碘化亚铜,1,10
‑
菲啰啉,碳酸铯,n,n
‑
二甲基甲酰胺,n2,100℃;(iii)对苯二胺,hatu,diea,二氯甲烷,室温;(iv)r3‑
cocl,三乙胺,二氯甲烷,0℃;(v)硒氰酸钾,碘化亚铜,1,10
‑
菲啰啉,碳酸铯,n,n
‑
二甲基甲酰胺,n2,100℃;(vi)对苯二胺,hatu,diea,二氯甲烷,室温;(vii)硒氰酸钾,碘化亚铜,1,10
‑
菲啰啉,碳酸铯,n,n
‑
二甲基甲酰胺,n2,100℃;(viii)氯化亚锡,无水乙醇,n2,室温;(ix)r3‑
cocl,三乙胺,二氯甲烷,0℃;(x)硒氰酸钾,碘化亚铜,1,10
‑
菲啰啉,碳酸铯,n,n
‑
二甲基甲酰胺,n2,100℃。
[0030]
其中,r1如上述通式i中所述;r3为二甲基胺磺酰基、环丙烷酰基、环丙烷磺酰基、4
‑
(二甲氨基)苯甲酰基。
[0031]
本发明所述的室温为20
‑
30℃。
[0032]
根据本发明优选的,含有苯并异硒唑酮衍生物的制备方法,具体步骤如下:
[0033]
(1)将邻碘苯甲酸(i
‑
1)、1h
‑
苯并三唑
‑1‑
基氧三吡咯烷基六氟磷酸盐加入到二氯甲烷中,随后向反应液中加入n,n
‑
二异丙基乙胺和取代胺,室温搅拌7~8小时,tlc监测;反应完毕,减压蒸除溶剂,然后向瓶内残留物中加入饱和碳酸氢钠溶液,二氯甲烷萃取,分取有机相,加入1n hcl溶液洗涤,分取有机相,加入饱和氯化钠溶液洗涤,有机相用无水硫酸钠干燥,过滤,滤液减压浓缩,所得粗品经硅胶柱层析分离纯化后得到中间体i
‑
2a~h;
[0034]
(2)将中间体i
‑
2a~h、硒氰酸钾、碘化亚铜、1,10
‑
菲啰啉、碳酸铯溶于n,n
‑
二甲基甲酰胺,n2保护,100℃加热回流,tlc监测;反应完毕,加入水,乙酸乙酯萃取,取有机相,加入饱和氯化钠溶液洗涤,有机相用无水硫酸钠干燥,过滤,滤液减压浓缩,所得粗品经硅胶柱层析分离纯化后得到目标产物i
‑
3~i
‑
10;
[0035]
(3)将邻碘苯甲酸(i
‑
1)、1h
‑
苯并三唑
‑1‑
基氧三吡咯烷基六氟磷酸盐加入到二氯甲烷中,随后向反应液中加入n,n
‑
二异丙基乙胺和对苯二胺,室温搅拌7
‑
8小时,tlc监测;反应完毕,减压蒸除溶剂,然后向瓶内残留物中加入饱和碳酸氢钠溶液,二氯甲烷萃取,分取有机相,加入1n hcl溶液洗涤,分取有机相,加入饱和氯化钠溶液洗涤,有机相用无水硫酸钠干燥,过滤,滤液减压浓缩,所得粗品经硅胶柱层析分离纯化后得到中间体i
‑
11;
[0036]
(4)将中间体i
‑
11、三乙胺加入到二氯甲烷中,冰浴条件下缓慢加入酰氯片段的二氯甲烷溶液,冰浴条件下反应5
‑
6小时,tlc检测;反应完毕,减压蒸除溶剂,然后向瓶内残留物中加入1n hcl溶液洗涤,二氯甲烷萃取,分取有机相,加入饱和氯化钠溶液洗涤,有机相用无水硫酸钠干燥,过滤,滤液减压浓缩,所得粗品经硅胶柱层析分离纯化后得到中间体i
‑
12a~d;
[0037]
(5)将中间体i
‑
12a~d、硒氰酸钾、碘化亚铜、1,10
‑
菲啰啉、碳酸铯溶于n,n
‑
二甲基甲酰胺,n2保护下,100℃加热回流,tlc监测;反应完毕,加入水,乙酸乙酯萃取,取有机相,加入饱和氯化钠溶液洗涤,有机相用无水硫酸钠干燥,过滤,滤液减压浓缩,所得粗品经硅胶柱层析分离纯化后得到目标产物i
‑
13~i
‑
16;
[0038]
(6)将2
‑
碘
‑4‑
硝基苯甲酸(i
‑
17)、1h
‑
苯并三唑
‑1‑
基氧三吡咯烷基六氟磷酸盐加入到二氯甲烷中,随后向反应液中加入n,n
‑
二异丙基乙胺和对苯二胺,室温搅拌7
‑
8小时,tlc监测;反应完毕,减压蒸除溶剂,然后向瓶内残留物中加入饱和碳酸氢钠溶液,二氯甲烷萃取,分取有机相,加入1n hcl溶液洗涤,分取有机相,加入饱和氯化钠溶液洗涤,有机相用无水硫酸钠干燥,过滤,滤液减压浓缩,所得粗品经硅胶柱层析分离纯化后得到中间体i
‑
18;
[0039]
(7)将中间体i
‑
18、硒氰酸钾、碘化亚铜、1,10
‑
菲啰啉、碳酸铯溶于n,n
‑
二甲基甲酰胺,n2保护下,100℃加热回流,tlc监测;反应完毕,加入水,乙酸乙酯萃取,取有机相,加入饱和氯化钠溶液洗涤,有机相用无水硫酸钠干燥,过滤,滤液减压浓缩,所得粗品经硅胶柱层析分离纯化后得到目标产物i
‑
19;
[0040]
(8)将i
‑
19与氯化亚锡溶于无水乙醇中,n2保护,室温下搅拌,tlc监测;反应完毕,加入氢氧化钠调节ph,过滤,减压蒸馏滤液,所得粗品经硅胶柱层析分离纯化后得到目标产物i
‑
20;
[0041]
(9)将中间体i
‑
18、三乙胺加入到二氯甲烷中,冰浴条件下缓慢加入含酰氯的二氯甲烷溶液,冰浴条件下反应5
‑
6小时,tlc检测;反应完毕,减压蒸除溶剂,然后向瓶内残留物中加入1n hcl溶液洗涤,二氯甲烷萃取,分取有机相,加入饱和氯化钠溶液洗涤,有机相用
无水硫酸钠干燥,过滤,滤液减压浓缩,所得粗品经硅胶柱层析分离纯化后得到中间体i
‑
21;
[0042]
(10)将中间体i
‑
21、硒氰酸钾、碘化亚铜、1,10
‑
菲啰啉、碳酸铯溶于n,n
‑
二甲基甲酰胺,n2保护,100℃加热回流,tlc监测;反应完毕,加入水,乙酸乙酯萃取,取有机相,加入饱和氯化钠溶液洗涤,有机相用无水硫酸钠干燥,过滤,滤液减压浓缩,所得粗品经硅胶柱层析分离纯化后得到目标产物i
‑
22。
[0043]
3.苯并异硒唑酮类衍生物的应用
[0044]
本发明公开了含苯并异硒唑酮衍生物抗主蛋白酶的筛选结果及其作为主蛋白酶抑制剂的应用。通过实验证明本发明的苯并异硒唑酮衍生物可作为主蛋白酶抑制剂用于制备抗冠状病毒药物。本发明还提供上述化合物在制备抗冠状病毒药物中的应用。
[0045]
本发明的一类含有苯并异硒唑酮衍生物可作为冠状病毒主蛋白酶抑制剂用于制备抗冠状病毒药物。
[0046]
一种抗冠状病毒药物组合物,包括本发明的一类含有苯并异硒唑酮衍生物和一种或多种药学上可接受载体或赋形剂。
[0047]
根据本发明优选的,所述的抗冠状病毒为sars
‑
cov
‑
2 m
pro
。
[0048]
本发明提供了一类含有苯并异硒唑酮衍生物及其制备方法,并通过酶活测试实验得出该系列化合物的主蛋白酶的酶抑制活性。具体地说,本发明在依布硒啉的基础上通过结构优化发现了活性更高的主蛋白酶抑制剂。
[0049]
说明书附图
[0050]
图1是以sars
‑
cov
‑
2 m
pro
为靶点的药物筛选原理图。
具体实施方式
[0051]
通过下述实施例有助于理解本发明,但是不能限制本发明的内容,所述百分比数均为质量百分比。
[0052]
实施例1:2
‑
碘
‑
n
‑
(萘
‑2‑
基甲基)苯甲酰胺(i
‑
2a)的制备
[0053]
将起始原料邻碘苯甲酸(i
‑
1)(0.50g,2.02mmol,1.0eq.)、1h
‑
苯并三唑
‑1‑
基氧三吡咯烷基六氟磷酸盐(1.15g,3.03mmol,1.5eq.)加入到20ml的二氯甲烷中,冰浴条件下搅拌30min;然后加入n,n
‑
二异丙基乙胺(1.01ml,6.06mmol,3eq.)和萘
‑2‑
基甲胺(0.48g,3.03mmol,1.5eq.),撤去冰浴转入室温搅拌,tlc监测;6h后反应完毕,减压蒸除溶剂,然后向瓶内残留物中加入饱和碳酸氢钠溶液40ml,二氯甲烷40ml萃取,分取有机相,加入1n hcl溶液40ml洗涤,分取有机相,加入饱和氯化钠溶液40ml洗涤,有机相用无水硫酸钠干燥,过滤,滤液减压浓缩,所得粗品经硅胶柱层析分离(洗脱剂乙酸乙酯:石油醚=1:8)得到中间体(s)
‑
(1
‑
((4
‑
甲氧基苯基)(甲基)氨基)
‑1‑
氧代
‑3‑
苯基丙烷
‑2‑
基)氨基甲酸叔丁酯(i
‑
2a)的粗品0.62g,白色固体,产率80%。
[0054]
波谱数据:1h nmr(400mhz,dmso
‑
d6)δ9.02(t,j=6.1hz,1h),7.91(d,j=8.0hz,4h),7.57(dd,j=8.6,1.6hz,1h),7.54
–
7.47(m,2h),7.46(dd,j=7.4,1.2hz,1h),7.41(dd,j=7.6,1.8hz,1h),7.19(td,j=7.5,1.8hz,1h),4.62(d,j=6.0hz,2h).esi
‑
ms:m/z 385.94(m
‑
1).c
18
h
14
ino[387.01].
[0055]
实施例2:2
‑
(萘
‑2‑
基甲基)苯并[d][1,2]硒唑
‑
3(2h)
‑
酮(i
‑
3)的制备
[0056]
将中间体i
‑
2a(0.40g,1.07mmol,1.0eq.)、硒氰酸钾(0.19g,2.57mmol,1.32eq.)、碘化亚铜(0.21g,1.10mmol,1.0eq.)、1,10
‑
菲啰啉(0.20g,1.11mmol,1.0eq.)、碳酸铯(0.87g,2.67mmol,2.5eq.)溶于10ml n,n
‑
二甲基甲酰胺,n2保护,100℃加热回流,tlc监测;反应完毕,加入20ml水,乙酸乙酯(3
×
10ml)萃取,取有机相,加入饱和氯化钠溶液(3
×
10ml)洗涤,有机相用无水硫酸钠干燥,过滤,滤液减压浓缩,所得粗品经硅胶柱层析分离纯化后得到目标产物2
‑
(萘
‑2‑
基甲基)苯并[d][1,2]硒唑
‑
3(2h)
‑
酮(i
‑
3)0.22g,白色固体,产率60%,熔点:188
‑
190℃。
[0057]
波谱数据:
[0058]1h nmr(400mhz,dmso
‑
d6)δ8.01(d,j=8.0hz,1h,phh),7.91(d,j=8.6hz,4h,phh),7.87(d,j=5.3hz,1h,phh),7.62(t,j=8.1hz,1h,phh),7.55
–
7.50(m,2h,phh),7.50
–
7.44(m,2h,phh),5.10(s,2h,ch2).
[0059]
13
c nmr(151mhz,dmso
‑
d6)δ166.92,139.83,136.47,133.34,132.86,132.08,128.73,128.33,128.17,128.05,128.01,126.93,126.82,126.66,126.52,126.34,47.46.
[0060]
esi
‑
ms:m/z 384.72(m+2na).c
14
h9f2nose[324.20].
[0061]
实施例3:n
‑
((1h
‑
吲哚
‑5‑
基)甲基)
‑2‑
碘苯甲酰胺(i
‑
2b)的制备
[0062]
将原料邻碘苯甲酸i
‑
1(0.50g,2.02mmol,1.0eq.)、1h
‑
苯并三唑
‑1‑
基氧三吡咯烷基六氟磷酸盐(1.15g,3.03mmol,1.5eq.)加入到20ml的二氯甲烷中,冰浴条件下搅拌30min;然后加入n,n
‑
二异丙基乙胺(1.01ml,6.06mmol,3.0eq.)和萘
‑2‑
基甲胺(0.44g,3.03mmol,1.5eq.),撤去冰浴转入室温搅拌,tlc监测;6h后反应完毕,减压蒸除溶剂,然后向瓶内残留物中加入饱和碳酸氢钠溶液40ml,二氯甲烷40ml萃取,分取有机相,加入1n hcl溶液40ml洗涤,分取有机相,加入饱和氯化钠溶液40ml洗涤,有机相用无水硫酸钠干燥,过滤,滤液减压浓缩,所得粗品经硅胶柱层析分离(洗脱剂ea:pe=1:4)得到中间体n
‑
((1h
‑
吲哚
‑5‑
基)甲基)
‑2‑
碘苯甲酰胺的粗品0.58g,白色固体,产率80%。
[0063]
波谱数据:1h nmr(400mhz,dmso
‑
d6)δ11.03(s,1h,nh),8.84(t,j=5.8hz,1h,nh),8.02
–
7.83(m,2h,phh),7.55(s,1h,phh),7.44(t,j=7.4hz,1h,phh),7.37
–
7.32(m,2h,phh),7.21
–
7.07(m,2h,phh),6.41(s,1h,phh),4.51(d,j=5.9hz,2h,ch2).esi
‑
ms:m/z 422.91(m+2na).c
16
h
13
in2o[376.19].
[0064]
实施例4:2
‑
((1h
‑
吲哚
‑5‑
基)甲基)苯并[d][1,2]硒唑
‑
3(2h)
‑
酮(i
‑
4)的制备
[0065]
将中间体i
‑
2b(0.80g,2.21mmol,1.0eq.)、硒氰酸钾(0.38g,2.65mmol,1.2eq.)、碘化亚铜(0.42g,2.21mmol,1.0eq.)、1,10
‑
菲啰啉(0.40g,2.21mmol,1.0eq.)、碳酸铯(1.80g,5.53mmol,2.5eq.)溶于10ml n,n
‑
二甲基甲酰胺,n2保护,100℃加热回流,tlc监测;反应完毕,加入20ml水,乙酸乙酯(3
×
10ml)萃取,取有机相,加入饱和氯化钠溶液(3
×
10ml)洗涤,有机相用无水硫酸钠干燥,过滤,滤液减压浓缩,所得粗品经硅胶柱层析分离纯化后得到目标产物2
‑
((1h
‑
吲哚
‑5‑
基)甲基)苯并[d][1,2]硒唑
‑
3(2h)
‑
酮(i
‑
4)0.43g,白色固体,产率60%,熔点:168
‑
170℃。
[0066]
波谱数据:1h nmr(400mhz,dmso
‑
d6)δ8.02(d,j=8.1hz,1h,nh),7.95
–
7.87(m,4h,phh),7.86(s,1h,indole
‑
ch),7.62(t,j=7.6hz,1h,indole
‑
ch),7.53
–
7.45(m,3h,phh),5.10(s,2h,ch2).
13
c nmr(151mhz,dmso
‑
d6)δ166.94,139.82,136.45,133.34,132.87,132.08,128.74,128.32,128.16,128.05,128.02,126.94,126.82,126.65,126.53,126.35,
126.33,47.48.
[0067]
esi
‑
ms:m/z 326.19(m
‑
1)c
16
h
12
n2ose[327.25].
[0068]
实施例5:2
‑
碘
‑
n
‑
(对甲苯基)苯甲酰胺(i
‑
2c)的制备
[0069]
将起始原料邻碘苯甲酸i
‑
1(0.60g,2.42mmol,1.0eq.)、1h
‑
苯并三唑
‑1‑
基氧三吡咯烷基六氟磷酸盐(1.38g,3.63mmol,1.5eq.)加入到20ml的二氯甲烷中,冰浴条件下搅拌30min;然后加入n,n
‑
二异丙基乙胺(1.20ml,7.26mmol,3.0eq.)和对甲基苯胺(0.44g,3.63mmol,1.5eq.),撤去冰浴转入室温搅拌,tlc监测;6h后反应完毕,减压蒸除溶剂,然后向瓶内残留物中加入40ml饱和碳酸氢钠溶液,40ml二氯甲烷萃取,分取有机相,加入1n hcl溶液40ml洗涤,分取有机相,加入40ml饱和氯化钠溶液洗涤,有机相用无水硫酸钠干燥,过滤,滤液减压浓缩,所得粗品经硅胶柱层析分离(洗脱剂乙酸乙酯:石油醚=1:2)得到中间体2
‑
碘
‑
n
‑
(对甲苯基)苯甲酰胺(i
‑
2c)的粗品0.66g,白色固体,产率80%。
[0070]
实施例6:2
‑
(对甲苯基)苯并[d][1,2]硒唑
‑
3(2h)
‑
酮(i
‑
5)的制备
[0071]
将中间体i
‑
2c(0.70g,1.99mmol,1.0eq.)、硒氰酸钾(0.34g,2.39mmol,1.2eq.)、碘化亚铜(0.38g,1.99mmol,1.0eq.)、1,10
‑
菲啰啉(0.36g,1.99mmol,1.0eq.)、碳酸铯(1.62g,4.98mmol,2.5eq.)溶于10ml n,n
‑
二甲基甲酰胺,n2保护,100℃加热回流,tlc监测;反应完毕,加入20ml水,乙酸乙酯(3
×
10ml)萃取,取有机相,加入饱和氯化钠溶液(3
×
10ml)洗涤,有机相用无水硫酸钠干燥,过滤,滤液减压浓缩,所得粗品经硅胶柱层析分离纯化后得到目标产物2
‑
(对甲苯基)苯并[d][1,2]硒唑
‑
3(2h)
‑
酮0.42g,白色固体,产率70%,熔点:140
‑
142℃。
[0072]
波谱数据:1h nmr(600mhz,dmso
‑
d6):δ8.01
–
7.98(m,1h,phh),7.86
–
7.83(m,1h,phh),7.62
–
7.57(m,1h,phh),7.43(t,j=7.5hz,1h,phh),7.22(d,j=8.0hz,2h,phh),7.16(d,j=7.9hz,2h,phh),2.28(s,3h,ch3).
13
c nmr(151mhz,dmso
‑
d6)δ166.75,139.73,137.23,135.76,131.98,129.56,129.37,128.51,128.39,127.93,126.28,21.17.esi
‑
ms:m/z 289.18(m+1).c
14
h
11
nose[288.21].
[0073]
实施例7:(4
‑
(2
‑
碘苯甲酰胺基)苯基)氨基甲酸叔丁酯(i
‑
2d)的制备
[0074]
将起始原料邻碘苯甲酸i
‑
1(0.60g,2.42mmol,1.0eq.)、1h
‑
苯并三唑
‑1‑
基氧三吡咯烷基六氟磷酸盐(1.38g,3.63mmol,1.5eq.)加入到20ml的二氯甲烷中,冰浴条件下搅拌30min;然后加入n,n
‑
二异丙基乙胺(1.20ml,7.26mmol,3.0eq.)和叔丁基(4
‑
氨基苯基)氨基甲酸酯(0.76g,3.63mmol,1.5eq.),撤去冰浴转入室温搅拌,tlc监测;6h后反应完毕,减压蒸除溶剂,然后向瓶内残留物中加入饱和碳酸氢钠溶液40ml,二氯甲烷40ml萃取,分取有机相,加入1n hcl溶液40ml洗涤,分取有机相,加入饱和氯化钠溶液40ml洗涤,有机相用无水硫酸钠干燥,过滤,滤液减压浓缩,所得粗品经硅胶柱层析分离(洗脱剂乙酸乙酯:石油醚=1:2)得到中间体2
‑
碘
‑
n
‑
(对甲苯基)苯甲酰胺(i
‑
2d)的粗品0.95g,白色固体,产率90%。
[0075]
波谱数据:esi
‑
ms:m/z 439.50(m+1).c
18
h
19
in2o3[438.26].
[0076]
实施例8:叔丁基(4
‑
(3
‑
氧代苯并[d][1,2]偶氮硒
‑
2(3h)
‑
基)苯基)氨基甲酸酯(i
‑
6)的制备
[0077]
将中间体i
‑
2d(0.40g,0.91mmol,1.0eq.)、硒氰酸钾(0.16g,1.10mmol,1.2eq.)、碘化亚铜(0.17g,0.91mmol,1.0eq.)、1,10
‑
菲啰啉(0.16g,0.91mmol,1.0eq.)、碳酸铯(0.16g,2.28mmol,2.5eq.)溶于10ml n,n
‑
二甲基甲酰胺,n2保护,100℃加热回流,tlc监
测;反应完毕,加入20ml水,乙酸乙酯(3
×
10ml)萃取,取有机相,加入饱和氯化钠溶液(3
×
10ml)洗涤,有机相用无水硫酸钠干燥,过滤,滤液减压浓缩,所得粗品经硅胶柱层析分离纯化后得到目标产物叔丁基(4
‑
(3
‑
氧代苯并[d][1,2]偶氮硒
‑
2(3h)
‑
基)苯基)氨基甲酸酯0.04g,棕色固体,产率10%,熔点:190
‑
192℃。
[0078]
波谱数据:1h nmr(400mhz,dmso
‑
d6):δ9.47(s,1h,nh),8.08(d,j=8.0hz,1h,phh),7.93
–
7.86(m,1h,phh),7.73
–
7.64(m,1h,phh),7.51(q,j=8.5,7.8hz,5h,phh),1.50(s,9h,ch3).
13
c nmr(150mhz,dmso
‑
d6):δ165.41,153.27,139.39,137.95,134.19,132.50,128.91,128.32,126.63,126.28,125.82,119.12,114.47,79.65,28.61.esi
‑
ms:m/z391.06(m+h).c
18
h
18
n2o3se[390.04].
[0079]
实施例9:4
‑
(2
‑
碘苯甲酰胺基)苯甲酸乙酯(i
‑
2e)的制备
[0080]
将起始原料邻碘苯甲酸(i
‑
1)(0.80g,3.23mmol,1.0eq.)、1h
‑
苯并三唑
‑1‑
基氧三吡咯烷基六氟磷酸盐(1.85g,4.85mmol,1.5eq.)加入到20ml的二氯甲烷中,冰浴条件下搅拌30min;然后加入n,n
‑
二异丙基乙胺(1.61ml,9.69mmol,3.0eq.)和4
‑
氨基苯甲酸乙酯(0.80g,4.85mmol,1.5eq.),撤去冰浴转入室温搅拌,tlc监测;6h后反应完毕,减压蒸除溶剂,然后向瓶内残留物中加入饱和碳酸氢钠溶液40ml,二氯甲烷40ml萃取,分取有机相,加入1n hcl溶液40ml洗涤,分取有机相,加入饱和氯化钠溶液40ml洗涤,有机相用无水硫酸钠干燥,过滤,滤液减压浓缩,所得粗品经硅胶柱层析分离(洗脱剂甲醇:二氯甲烷=1:50)得到中间体4
‑
(2
‑
碘苯甲酰胺基)苯甲酸乙酯(i
‑
2e)的粗品0.93g,白色固体,产率73%。
[0081]
波谱数据:esi
‑
ms:m/z 396.07(m+h).c
16
h
14
ino3[395.19].
[0082]
实施例10:4
‑
(3
‑
氧代苯并[d][1,2]偶氮硒
‑
2(3h)
‑
基)苯甲酸乙酯(i
‑
7)的制备
[0083]
将中间体i
‑
2e(0.27g,0.68mmol,1.0eq.)、硒氰酸钾(0.12g,0.82mmol,1.2eq.)、碘化亚铜(0.13g,0.68mmol,1.0eq.)、1,10
‑
菲啰啉(0.12g,0.68mmol,1.0eq.)、碳酸铯(0.55g,1.70mmol,2.5eq.)溶于10ml n,n
‑
二甲基甲酰胺,n2保护,100℃加热回流,tlc监测;反应完毕,加入20ml水,乙酸乙酯(3
×
10ml)萃取,取有机相,加入饱和氯化钠溶液(3
×
10ml)洗涤,有机相用无水硫酸钠干燥,过滤,滤液减压浓缩,所得粗品经硅胶柱层析分离纯化后得到目标产物4
‑
(3
‑
氧代苯并[d][1,2]偶氮硒
‑
2(3h)
‑
基)苯甲酸甲酯0.06g,淡黄色固体,产率26%,熔点:180
‑
182℃。
[0084]
波谱数据:1h nmr(400mhz,dmso
‑
d6)δ9.47(s,1h,nh),8.08(d,j=8.0hz,1h,phh),7.93
–
7.86(m,1h,phh),7.73
–
7.64(m,1h,phh),7.51(q,j=8.5,7.8hz,5h,phh),1.50(s,9h,ch3).
13
c nmr(150mhz,dmso
‑
d6)δ165.41,153.27,139.39,137.95,134.19,132.50,128.91,128.32,126.63,126.28,125.82,119.12,114.47,79.65,28.61.esi
‑
ms:m/z348.11(m+1).c
16
h
13
no3se[347.00].
[0085]
实施例11:n
‑
(3,4
‑
二氯苯基)
‑2‑
碘苯甲酰胺(i
‑
2f)的制备
[0086]
将起始原料邻碘苯甲酸i
‑
1(0.80g,3.23mmol,1.0eq.)、1h
‑
苯并三唑
‑1‑
基氧三吡咯烷基六氟磷酸盐(1.85g,4.85mmol,1.5eq.)加入到10ml的二氯甲烷中,冰浴条件下搅拌30min;然后加入n,n
‑
二异丙基乙胺(0.17ml,9.69mmol,3.0eq.)和3,4
‑
二氯苯胺(0.79g,4.85mmol,1.5eq.),撤去冰浴转入室温搅拌,tlc监测;6h后反应完毕,减压蒸除溶剂,然后向瓶内残留物中加入饱和碳酸氢钠溶液40ml,二氯甲烷40ml萃取,分取有机相,加入1n hcl溶液40ml洗涤,分取有机相,加入饱和氯化钠溶液40ml洗涤,有机相用无水硫酸钠干燥,过
滤,滤液减压浓缩,所得粗品经硅胶柱层析分离(洗脱剂乙酸乙酯:石油醚=1:2)得到中间体n
‑
(3,4
‑
二氯苯基)
‑2‑
碘苯甲酰胺(i
‑
2f)的粗品0.86g,白色固体,产率68%。
[0087]
波谱数据:1h nmr(400mhz,dmso
‑
d6):δ10.73(s,1h,nh),8.09(d,j=1.4hz,1h,phh),7.95(d,j=7.9hz,1h,phh),7.63(d,j=1.4hz,2h,phh),7.51(q,j=4.6,3.8hz,2h,phh),7.25(ddd,j=7.9,5.9,3.3hz,1h,phh).esi
‑
ms:m/z 389.99(m
‑
1).c
13
h8c
l2
ino[390.90].
[0088]
实施例12:2
‑
(3,4
‑
二氯苯基)苯并[d][1,2]硒唑
‑
3(2h)
‑
酮(i
‑
8)的制备
[0089]
将中间体i
‑
2e(0.30g,0.91mmol,1.0eq.)、硒氰酸钾(0.16g,1.10mmol,1.2eq.)、碘化亚铜(0.17g,0.91mmol,1.0eq.)、1,10
‑
菲啰啉(0.16g,0.91mmol,1.0eq.)、碳酸铯(0.74g,2.28mmol,2.5eq.)溶于10ml n,n
‑
二甲基甲酰胺,n2保护,100℃加热回流,tlc监测;反应完毕,加入20ml水,乙酸乙酯(3
×
10ml)萃取,取有机相,加入饱和氯化钠溶液(3
×
10ml)洗涤,有机相用无水硫酸钠干燥,过滤,滤液减压浓缩,所得粗品经硅胶柱层析分离纯化后得到目标产物叔丁基(4
‑2‑
(3,4
‑
二氯苯基)苯并[d][1,2]硒唑
‑
3(2h)
‑
酮0.23g,浅棕色固体,产率72%,熔点:190
‑
192℃。
[0090]
波谱数据:1h nmr(400mhz,dmso
‑
d6):δ8.14
–
8.05(m,2h),7.92(d,j=7.7hz,1h,phh),7.70(t,j=8.3hz,2h,phh),7.62
–
7.56(m,1h,phh),7.49(t,j=7.5hz,1h,phh).
13
c nmr(150mhz,dmso
‑
d6):δ165.92,140.35,139.23,133.12,131.86,131.41,128.63,128.56,128.01,126.88,126.34,126.12,124.79.esi
‑
ms:m/z 344.08(m+1).c
13
h7c
l2
nose[343.07].
[0091]
实施例13:n
‑
(3,5
‑
二氟苄基)
‑2‑
碘苯甲酰胺(i
‑
2g)的制备
[0092]
将起始原料邻碘苯甲酸i
‑
1(0.60g,2.42mmol,1.0eq.)、1h
‑
苯并三唑
‑1‑
基氧三吡咯烷基六氟磷酸盐(1.38g,3.63mmol,1.5eq.)加入到10ml的二氯甲烷中,冰浴条件下搅拌30min;然后加入n,n
‑
二异丙基乙胺(1.20ml,7.26mmol,3.0eq.)和叔丁基(4
‑
氨基苯基)氨基甲酸酯(0.76g,3.63mmol,1.5eq.),撤去冰浴转入室温搅拌,tlc监测;6h后反应完毕,减压蒸除溶剂,然后向瓶内残留物中加入30ml饱和碳酸氢钠溶液,用30ml二氯甲烷萃取,分取有机相,加入30ml1n hcl溶液洗涤,分取有机相,加入30ml饱和氯化钠溶液洗涤,有机相用无水硫酸钠干燥,过滤,滤液减压浓缩,所得粗品经硅胶柱层析分离(洗脱剂甲醇:二氯甲烷=1:50)得到中间体n
‑
(3,5
‑
二氟苄基)
‑2‑
碘苯甲酰胺(i
‑
2g)的粗品0.58g,白色固体,产率64%。
[0093]
波谱数据:esi
‑
ms:m/z 371.96(m
‑
1).c
14
h
10
f2ino[372.97].
[0094]
实施例14:2
‑
(3,5
‑
二氟苄基)苯并[d][1,2]硒唑
‑
3(2h)
‑
酮(i
‑
9)的制备
[0095]
将中间体i
‑
2g(0.40g,1.07mmol,1.0eq.)、硒氰酸钾(0.18g,1.28mmol,1.2eq.)、碘化亚铜(0.20g,1.07mmol,1.0eq.)、1,10
‑
菲啰啉(0.19g,1.07mmol,1.0eq.)、碳酸铯(0.87g,2.68mmol,2.5eq.)溶于10ml n,n
‑
二甲基甲酰胺,n2保护,100℃加热回流,tlc监测;反应完毕,加入20ml水,乙酸乙酯(3
×
10ml)萃取,取有机相,加入饱和氯化钠溶液(3
×
10ml)洗涤,有机相用无水硫酸钠干燥,过滤,滤液减压浓缩,所得粗品经硅胶柱层析分离纯化后得到目标产物叔丁基(4
‑2‑
(3,5
‑
二氟苄基)苯并[d][1,2]硒唑
‑
3(2h)
‑
酮0.04g,白色固体,产率11%,熔点:144
‑
146℃。
[0096]
波谱数据:1h nmr(400mhz,dmso
‑
d6):δ8.02(d,j=8.0hz,1h,phh),7.85(d,j=7.6hz,1h,phh),7.66
–
7.59(m,1h,phh),7.41(dd,j=12.0,7.0hz,2h,phh),7.29(td,j=
10.1,2.5hz,1h,phh),7.09(td,j=8.5,2.1hz,1h,phh),4.94(s,2h,ch2).
13
c nmr(150mhz,dmso
‑
d6):δ166.85,139.73,132.16,129.67,128.08,127.95,126.38,126.36,121.97(dd,j=15.3,3.6hz),112.11(dd,j=21.3,3.6hz),104.42(t,j=25.8hz),40.72.esi
‑
ms:m/z325.76(m+1),347.77(m+na).c
14
h9f2nose[324.20].
[0097]
实施例15:叔丁基(4
‑
(2
‑
碘苯甲酰胺基)苄基)氨基甲酸酯(i
‑
2h)的制备
[0098]
将起始原料邻碘苯甲酸i
‑
1(0.60g,2.42mmol,1.0eq.)、1h
‑
苯并三唑
‑1‑
基氧三吡咯烷基六氟磷酸盐(1.38g,3.63mmol,1.5eq.)加入到20ml的二氯甲烷中,冰浴条件下搅拌30min;然后加入n,n
‑
二异丙基乙胺(1.20ml,7.26mmol,3.0eq.)和叔丁基(4
‑
氨基苯基)氨基甲酸酯(0.76g,3.63mmol,1.5eq.),撤去冰浴转入室温搅拌,tlc监测;6h后反应完毕,减压蒸除溶剂,然后向瓶内残留物中加入饱和碳酸氢钠溶液40ml,二氯甲烷40ml萃取,分取有机相,加入1n hcl溶液40ml洗涤,分取有机相,加入饱和氯化钠溶液40ml洗涤,有机相用无水硫酸钠干燥,过滤,滤液减压浓缩,所得粗品经硅胶柱层析分离(洗脱剂乙酸乙酯:石油醚=1:2)得到中间体叔丁基(4
‑
(2
‑
碘苯甲酰胺基)苄基)氨基甲酸酯(i
‑
2h)的粗品0.95g,白色固体,产率90%。
[0099]
实施例16:叔丁基(4
‑
(3
‑
氧代苯并[d][1,2]偶氮硒
‑
2(3h)
‑
基)苄基)氨基甲酸酯(i
‑
10)的制备
[0100]
将中间体i
‑
2e(0.40g,0.88mmol,1.0eq.),硒氰酸钾(0.15g,1.06mmol,1.2eq.),碘化亚铜(0.17g,0.88mmol,1.0eq.),1,10
‑
菲啰啉(0.16g,0.88mmol,1.0eq.),碳酸铯(0.72g,2.20mmol,2.5eq.)溶于10ml n,n
‑
二甲基甲酰胺,n2保护,100℃加热回流,tlc监测;反应完毕,加入20ml水,乙酸乙酯(3
×
10ml)萃取,取有机相,加入饱和氯化钠溶液(3
×
10ml)洗涤,有机相用无水硫酸钠干燥,过滤,滤液减压浓缩,所得粗品经硅胶柱层析分离纯化后得到目标产物叔丁基(4
‑
(3
‑
氧代苯并[d][1,2]偶氮硒
‑
2(3h)
‑
基)苯基)氨基甲酸酯0.04g,白色固体,产率11%,熔点:182
‑
184℃。
[0101]
波谱数据:1h nmr(600mhz,dmso
‑
d6):δ8.08(d,j=8.1hz,1h,phh),7.90(d,j=7.7hz,1h,phh),7.71
–
7.65(m,1h),7.57(d,j=8.1hz,2h,phh),7.48(t,j=7.5hz,1h,phh),7.41(t,j=5.9hz,1h,nh),7.31(d,j=8.3hz,2h,phh),4.14(d,j=6.0hz,2h,ch2),1.41(s,9h,ch3).
13
c nmr(150mhz,dmso
‑
d6):δ165.47,156.28,139.36,138.73,138.42,132.66,128.94,128.39,128.19,126.70,126.28,125.10,114.16,78.31,43.49,28.75.esi
‑
ms:m/z404.56(m+1),426.74(m+na).c
19
h
20
n2o3se[403.35]
[0102]
实施例17:n
‑
(4
‑
氨基苯基)
‑2‑
碘苯甲酰胺(i
‑
11)的制备
[0103]
将原料邻碘苯甲酸i
‑
1(1.00g,4.03mmol,1.0eq.)、1h
‑
苯并三唑
‑1‑
基氧三吡咯烷基六氟磷酸盐(2.30g,6.05mmol,1.5eq.)、n,n
‑
二异丙基乙胺(2.00ml,12.09mmol,3.0eq.)和苯
‑
1,4
‑
二胺(0.65g,6.05mmol,1.5eq.)加入到15ml的n,n
‑
二甲基甲酰胺中,室温搅拌,tlc监测;6h后反应完毕,反应完毕,加入20ml水,乙酸乙酯(3
×
10m l)萃取,取有机相,加入饱和氯化钠溶液(3
×
10ml)洗涤,有机相用无水硫酸钠干燥,过滤,滤液减压浓缩,所得粗品经硅胶柱层析分离(洗脱剂甲醇:二氯甲烷=1:100)得到中间体n
‑
(4
‑
氨基苯基)
‑2‑
碘苯甲酰胺(i
‑
11)的粗品0.97g,淡黄色固体,产率72%。
[0104]
波谱数据:1h nmr(400mhz,dmso
‑
d6):δ9.95(s,1h),7.90(d,j=7.8hz,1h),7.55
–
7.40(m,2h),7.34(d,j=8.7hz,1h),7.19(td,j=7.8,1.7hz,1h),6.54(d,j=8.7hz,2h),
4.95(s,2h).esi
‑
ms:m/z 338.56(m+1).c
13
h
11
in2o[337.99].
[0105]
实施例18:n
‑
(4
‑
((n,n
‑
二甲基氨磺酰)氨基)苯基)
‑2‑
碘苯甲酰胺(i
‑
12a)的制备
[0106]
将中间体n
‑
(4
‑
氨基苯基)
‑2‑
碘苯甲酰胺(i
‑
11)(0.40g,1.18mmol,1.0eq.)溶于4ml的二氯甲烷中,加入三乙胺(0.2ml,1.42mmol,1.2eq.),冰浴条件下搅拌,随后将二甲基胺磺酰氯(0.2g,1.30mmol,1.1eq.)溶于2.0ml的二氯甲烷中,缓慢加入上述溶液中,0℃条件下搅拌,tlc监测;6h后反应完毕,反应完毕,加入饱和碳酸氢钠溶液(3
×
6.0ml),二氯甲烷30ml萃取,取有机相;加入饱和氯化钠溶液(3
×
10ml)洗涤,有机相用无水硫酸钠干燥,过滤,滤液减压浓缩,所得粗品经硅胶柱层析分离(洗脱剂甲醇:二氯甲烷=1:50)得到n
‑
(4
‑
((n,n
‑
二甲基氨磺酰)氨基)苯基)
‑2‑
碘苯甲酰胺(i
‑
12a)的粗品0.17g,淡黄色固体,产率32%。
[0107]
波谱数据:1h nmr(400mhz,dmso
‑
d6):δ10.36(s,1h),9.76(s,1h),7.93(d,j=7.9hz,1h),7.63(d,j=8.7hz,2h),7.53
–
7.43(m,2h),7.27
–
7.15(m,3h),2.69(s,6h).esi
‑
ms:m/z 444.08(m
‑
1).c
15
h
16
in3o3s[445.28].
[0108]
实施例19:n
‑
(4
‑
((n,n
‑
二甲基氨磺酰)氨基)苯基)
‑
苯并[d][1,2]硒唑
‑
3(2h)
‑
酮(i
‑
13)的制备
[0109]
将中间体i
‑
12a(0.15g,0.34mmol,1.0eq.)、硒氰酸钾(0.06g,0.41mmol,1.2eq.)、碘化亚铜(0.07g,0.34mmol,1.0eq.)、1,10
‑
菲啰啉(0.06g,0.34mmol,1.0eq.)、碳酸铯(0.28g,0.85mmol,2.5eq.)溶于6ml n,n
‑
二甲基甲酰胺,n2保护,100℃加热回流,tlc监测;反应完毕,加入6ml水,乙酸乙酯(3
×
10ml)萃取,取有机相,加入饱和氯化钠溶液(3
×
10ml)洗涤,有机相用无水硫酸钠干燥,过滤,滤液减压浓缩,所得粗品经硅胶柱层析(洗脱剂甲醇:二氯甲烷=1:30)分离纯化后得到目标产物n
‑
(4
‑
((n,n
‑
二甲基氨磺酰)氨基)苯基)
‑
苯并[d][1,2]硒唑
‑
3(2h)
‑
酮(i
‑
13)0.02g,黄色固体,产率11%,熔点:180
‑
182℃。
[0110]
波谱数据:1h nmr(400mhz,dmso
‑
d6):δ10.01(s,1h,nh),8.09(d,j=8.1hz,1h,phh),7.90(dd,j=7.8,1.4hz,1h,phh),7.73
–
7.64(m,1h,phh),7.60
–
7.53(m,2h,phh),7.48(t,j=7.6hz,1h,phh),7.32
–
7.23(m,2h,phh),2.74(s,6h,2ch3).
13
c nmr(100mhz,dmso
‑
d6):δ165.47,139.36,136.86,135.36,132.64,128.82,128.37,126.70,126.29,126.17,120.25,38.17.esi
‑
ms:m/z 397.80(m+1),419.86(m+na).c
15
h
15
n3o3sse[396.33].
[0111]
实施例20:n
‑
(4
‑
(环丙烷酰胺基)苯基)
‑2‑
碘苯甲酰胺(i
‑
12b)的制备
[0112]
将中间体n
‑
(4
‑
氨基苯基)
‑2‑
碘苯甲酰胺i
‑
11(0.20g,0.59mmol,1.0eq.)溶于4ml的二氯甲烷中,加入三乙胺(0.10ml,0.71mmol,1.2eq.),冰浴条件下搅拌,随后将环丙烷酰氯(0.07g,0.65mmol,1.1eq.)溶于2.0ml的二氯甲烷中,缓慢加入上述溶液中,0℃条件下搅拌,tlc监测;6h后反应完毕,反应完毕,加入饱和碳酸氢钠溶液(3
×
6.0ml),二氯甲烷20ml萃取,取有机相;加入饱和氯化钠溶液(3
×
10ml)洗涤,有机相用无水硫酸钠干燥,过滤,滤液减压浓缩,所得粗品经硅胶柱层析分离(洗脱剂甲醇:二氯甲烷=1:100)得到n
‑
(4
‑
(环丙烷酰胺基)苯基)
‑2‑
碘苯甲酰胺(i
‑
12b)的粗品0.12g,白色固体,产率48%。
[0113]
波谱数据:1h nmr(400mhz,dmso
‑
d6):δ10.32(s,1h),10.15(s,1h),7.92(d,j=7.9hz,1h),7.62(d,j=9.0hz,2h),7.55(d,j=8.8hz,2h),7.52
–
7.44(m,2h),7.22(td,j=7.4,2.1hz,1h),2.69(s,1h),0.78(t,j=6.6hz,4h).esi
‑
ms:m/z 429.04(m+na).c
17
h
15
in2o2[406.22].
[0114]
实施例21:n
‑
(4
‑
(3
‑
氧代苯并[d][1,2]偶氮硒
‑
2(3h)
‑
基)苯基)环丙烷甲酰胺(i
‑
14)的制备
[0115]
将中间体i
‑
12b(0.15g,0.37mmol,1.0eq.)、硒氰酸钾(0.06g,0.44mmol,1.2eq.)、碘化亚铜(0.07g,0.37mmol,1.0eq.)、1,10
‑
菲啰啉(0.07g,0.37mmol,1.0eq.)、碳酸铯(0.30g,0.93mmol,2.5eq.)溶于6ml n,n
‑
二甲基甲酰胺,n2保护,100℃加热回流,tlc监测;反应完毕,加入6ml水,乙酸乙酯(3
×
10ml)萃取,取有机相,加入饱和氯化钠溶液(3
×
10ml)洗涤,有机相用无水硫酸钠干燥,过滤,滤液减压浓缩,所得粗品经硅胶柱层析(洗脱剂甲醇:二氯甲烷=1:100)分离纯化后得到目标产物n
‑
(4
‑
(3
‑
氧代苯并[d][1,2]偶氮硒
‑
2(3h)
‑
基)苯基)环丙烷甲酰胺(i
‑
14)0.08g,白色固体,产率60%,熔点:183
‑
185℃。
[0116]
波谱数据:1h nmr(400mhz,dmso
‑
d6):δ10.31(s,1h),8.09(d,j=8.0hz,1h),7.90(d,j=7.6hz,1h),7.68(dd,j=7.3,5.2hz,3h),7.55(d,j=8.8hz,2h),7.48(t,j=7.5hz,1h),1.80(ddd,j=12.5,7.6,4.9hz,1h),0.87
–
0.77(m,4h).
13
c nmr(100mhz,dmso
‑
d6):δ172.10,165.40,139.36,137.68,134.88,132.58,128.92,128.35,126.68,126.30,125.72,119.96,15.03,7.71.esi
‑
ms:m/z 359.13(m+1).c
17
h
14
n2o2se[358.02].
[0117]
实施例22:n
‑
(4
‑
(环丙烷磺酰胺)苯基)
‑2‑
碘苯甲酰胺(i
‑
12c)的制备
[0118]
将中间体n
‑
(4
‑
氨基苯基)
‑2‑
碘苯甲酰胺(i
‑
11)(0.40g,1.18mmol,1.0eq.)溶于4ml的二氯甲烷中,加入三乙胺(0.19ml,1.42mmol,1.2eq.),冰浴条件下搅拌,随后将环丙烷磺酰氯(0.18g,1.30mmol,1.1eq.)溶于2.0ml的二氯甲烷中,缓慢加入上述溶液中,0℃条件下搅拌,tlc监测;6h后反应完毕,反应完毕,加入饱和碳酸氢钠溶液(3
×
6.0ml),二氯甲烷20ml萃取,取有机相;加入饱和氯化钠溶液(3
×
10ml)洗涤,有机相用无水硫酸钠干燥,过滤,滤液减压浓缩,所得粗品经硅胶柱层析分离(洗脱剂甲醇:二氯甲烷=1:50)得到n
‑
(4
‑
(环丙烷磺酰胺)苯基)
‑2‑
碘苯甲酰胺(i
‑
12c)的粗品0.13g,白色固体,产率25%。
[0119]
波谱数据:esi
‑
ms:m/z 440.10(m
‑
1).c
17
h
14
n2o2se[441.98].
[0120]
实施例23:n
‑
(4
‑
(3
‑
氧代苯并[d][1,2]偶氮硒
‑
2(3h)
‑
基)苯基)环丙烷磺酰胺(i
‑
15)的制备
[0121]
将中间体i
‑
12c(0.48g,0.98mmol,1.0eq.)、硒氰酸钾(0.17g,1.18mmol,1.2eq.)、碘化亚铜(0.19g,0.98mmol,1.0eq.)、1,10
‑
菲啰啉(0.18g,0.98mmol,1.0eq.)、碳酸铯(0.80g,2.45mmol,2.5eq.)溶于6ml n,n
‑
二甲基甲酰胺,n2保护,100℃加热回流,tlc监测;反应完毕,加入6ml水,乙酸乙酯(3
×
10ml)萃取,取有机相,加入饱和氯化钠溶液(3
×
10ml)洗涤,有机相用无水硫酸钠干燥,过滤,滤液减压浓缩,所得粗品经硅胶柱层析(洗脱剂甲醇:二氯甲烷=1:100)分离纯化后得到目标产物n
‑
(4
‑
(3
‑
氧代苯并[d][1,2]偶氮硒
‑
2(3h)
‑
基)苯基)环丙烷磺酰胺(i
‑
15)0.07g,棕色固体,产率18%,熔点:174
‑
176℃。
[0122]
波谱数据:1h nmr(400mhz,dmso
‑
d6):δ9.83(s,1h),8.09(d,j=8.1hz,1h),7.95(s,1h),7.91(d,j=7.7hz,1h),7.68(t,j=7.6hz,1h),7.60(d,j=8.6hz,1h),7.48(t,j=7.5hz,1h),7.32(d,j=8.7hz,2h),2.89(s,2h),2.73(s,2h),2.70
–
2.62(m,1h).
13
c nmr(100mhz,dmso
‑
d6):δ165.49,162.79,139.33,136.46,136.03,134.28,132.68,132.22,128.84,128.39,127.09,126.73,126.29,126.13,122.92,121.62,121.46,114.28,36.25,31.25,30.06,5.51,5.40.esi
‑
ms:m/z 416.90(m+na).c
17
h
14
n2o2se[393.33].
[0123]
实施例24:n
‑
(4
‑
(4
‑
(二甲氨基)苯甲酰胺基)苯基)
‑2‑
碘苯甲酰胺(i
‑
12d)的制备
[0124]
将中间体n
‑
(4
‑
氨基苯基)
‑2‑
碘苯甲酰胺(i
‑
11)(0.50g,1.48mmol,1.0eq.)溶于4ml的二氯甲烷中,加入三乙胺(0.25ml,1.78mmol,1.2eq.),冰浴条件下搅拌,随后将4
‑
(二甲氨基)苯甲酰氯(0.30g,1.63mmol,1.1eq.)溶于2.0ml的二氯甲烷中,缓慢加入上述溶液中,0℃条件下搅拌,tlc监测;6h后反应完毕,反应完毕,加入饱和碳酸氢钠溶液(3
×
6.0ml),二氯甲烷20ml萃取,取有机相;加入饱和氯化钠溶液(3
×
10ml)洗涤,有机相用无水硫酸钠干燥,过滤,滤液减压浓缩,所得粗品经硅胶柱层析分离(洗脱剂甲醇:二氯甲烷=1:50)得到n
‑
(4
‑
(4
‑
(二甲氨基)苯甲酰胺基)苯基)
‑2‑
碘苯甲酰胺(i
‑
12d)的粗品0.07g,白色固体,产率13%。
[0125]
波谱数据:1h nmr(400mhz,dmso
‑
d6):δ10.36(d,j=8.5hz,1h),10.16
–
9.86(m,1h),7.90(dd,j=22.1,8.4hz,2h),7.74(dd,j=8.7,6.6hz,1h),7.66(d,j=8.6hz,2h),7.55(s,1h),7.51
–
7.46(m,2h),7.20(dt,j=16.7,8.0hz,2h),6.73(dd,j=23.3,8.9hz,2h),3.00(d,j=6.0hz,6h).
[0126]
实施例25:n
‑
(4
‑
(4
‑
(二甲氨基)苯甲酰胺基)苯基)
‑2‑
碘苯甲酰胺(i
‑
16)的制备
[0127]
将中间体i
‑
12d(0.48g,0.98mmol,1.0eq.)、硒氰酸钾(0.17g,1.18mmol,1.2eq.)、碘化亚铜(0.19g,0.98mmol,1.0eq.)、1,10
‑
菲啰啉(0.18g,0.98mmol,1.0eq.)、碳酸铯(0.80g,2.45mmol,2.5eq.)溶于6ml n,n
‑
二甲基甲酰胺,n2保护,100℃加热回流,tlc监测;反应完毕,加入6ml水,乙酸乙酯(3
×
10ml)萃取,取有机相,加入饱和氯化钠溶液(3
×
10ml)洗涤,有机相用无水硫酸钠干燥,过滤,滤液减压浓缩,所得粗品经硅胶柱层析(洗脱剂甲醇:二氯甲烷=1:100)分离纯化后得到目标产物n
‑
(4
‑
(4
‑
(二甲氨基)苯甲酰胺基)苯基)
‑2‑
碘苯甲酰胺0.07g,棕色固体,产率18%,熔点:174
‑
176℃。
[0128]
波谱数据:1h nmr(600mhz,dmso
‑
d6):δ10.31(s,1h),8.31(d,j=1.8hz,1h),8.10(d,j=8.0hz,1h),7.95
–
7.89(m,1h),7.74
–
7.63(m,4h),7.63
–
7.53(m,3h),7.49(t,j=7.4hz,1h),7.29(d,j=8.8hz,1h),3.36(s,6h).
13
c nmr(100mhz,dmso
‑
d6)δ165.45,163.04,160.08,139.36,136.48,135.52,132.64,129.53,128.88,128.38,126.71,126.48,126.31,125.86,120.20,118.55,111.23,49.08.esi
‑
ms:m/z 453.69(m+nh
4+
).c
22
h
19
n3o2se[436.38].
[0129]
实施例26:n
‑
(4
‑
氨基苯基)
‑2‑
碘
‑4‑
硝基苯甲酰胺(i
‑
18)的制备
[0130]
将起始原料2
‑
碘
‑4‑
硝基苯甲酸(i
‑
17)(1.00g,3.41mmol,1.0eq.)、1h
‑
苯并三唑
‑1‑
基氧三吡咯烷基六氟磷酸盐(1.95g,5.12mmol,1.5eq.)、n,n
‑
二异丙基乙胺(1.70ml,10.23mmol,3.0eq.)和苯
‑
1,4
‑
二胺(0.55g,5.12mmol,1.5eq.)加入到20ml的n,n
‑
二甲基甲酰胺中,室温搅拌,tlc监测;6h后反应完毕,反应完毕,加入20ml水,乙酸乙酯(3
×
10ml)萃取,取有机相,加入饱和氯化钠溶液(3
×
10ml)洗涤,有机相用无水硫酸钠干燥,过滤,滤液减压浓缩,所得粗品经硅胶柱层析分离(洗脱剂甲醇:二氯甲烷=1:50)得到中间体n
‑
(4
‑
氨基苯基)
‑2‑
碘
‑4‑
硝基苯甲酰胺(i
‑
18)的粗品0.80g,淡黄色固体,产率62%。
[0131]
波谱数据:1h nmr(400mhz,dmso
‑
d6):δ10.18(s,1h),8.63(d,j=2.1hz,1h),8.30(dd,j=8.4,2.1hz,1h),7.69(d,j=8.4hz,1h),7.34(d,j=8.7hz,2h),6.56(d,j=8.6hz,2h),5.04(s,2h).
[0132]
实施例27:2
‑
(4
‑
氨基苯基)
‑6‑
硝基苯并[d][1,2]硒唑
‑
3(2h)
‑
酮(i
‑
19)的制备
[0133]
将中间体i
‑
18(0.13g,0.34mmol,1.0eq.)、硒氰酸钾(0.06g,0.41mmol,1.2eq.)、
碘化亚铜(0.06g,0.98mmol,1.0eq.)、1,10
‑
菲啰啉(0.06g,0.34mmol,1.0eq.)、碳酸铯(0.28g,0.85mmol,2.5eq.)溶于6ml n,n
‑
二甲基甲酰胺,n2保护,100℃加热回流,tlc监测;反应完毕,加入6ml水,乙酸乙酯(3
×
10ml)萃取,取有机相,加入饱和氯化钠溶液(3
×
10ml)洗涤,有机相用无水硫酸钠干燥,过滤,滤液减压浓缩,所得粗品经硅胶柱层析(洗脱剂甲醇:二氯甲烷=1:50)分离纯化后得到目标产物2
‑
(4
‑
氨基苯基)
‑6‑
硝基苯并[d][1,2]硒唑
‑
3(2h)
‑
酮(i
‑
19)0.01g,棕色固体,产率12%,熔点:220
‑
222℃。
[0134]
波谱数据:1h nmr(400mhz,dmso
‑
d6):δ9.88(s,1h),8.94(d,j=1.8hz,1h),8.26(dd,j=8.5,1.9hz,1h),8.09(d,j=8.5hz,1h),7.62(d,j=8.7hz,2h),7.34(d,j=8.7hz,2h),2.68(p,j=6.5hz,1h),0.98(d,j=6.3hz,4h).
13
c nmr(150mhz,dmso
‑
d6):δ163.66,149.92,148.32,140.57,133.69,129.33,127.50,126.93,121.98,121.39,114.30.esi
‑
ms:m/z 335.97(m+1).c
13
h9n3o3se[334.20].
[0135]
实施例28:6
‑
氨基
‑2‑
(4
‑
氨基苯基)苯并[d][1,2]硒唑
‑
3(2h)
‑
酮(i
‑
20)的制备
[0136]
将目标产物i
‑
19(0.20g,0.60mmol,1.0eq.)溶于10ml的无水乙醇中,随后加入氯化亚锡,n2保护下,室温搅拌,tlc监测;反应完毕,加入1m氢氧化钠溶液调节ph至7,将反应液过滤,乙酸乙酯洗涤残渣,洗液加入饱和氯化钠溶液15ml,乙酸乙酯(3
×
10ml)萃取,合并有机相,用无水硫酸钠干燥,过滤,滤液减压浓缩,所得粗品经硅胶柱层析(洗脱剂甲醇:二氯甲烷=1:50)分离纯化后得到目标产物6
‑
氨基
‑2‑
(4
‑
氨基苯基)苯并[d][1,2]硒唑
‑
3(2h)
‑
酮(i
‑
20)0.11g,棕色固体,产率11%,熔点:220
‑
222℃。
[0137]
波谱数据:1h nmr(400mhz,dmso
‑
d6):δ7.46(d,j=8.4hz,1h),7.10(d,j=8.5hz,2h),7.06(d,j=1.5hz,1h),6.65
–
6.52(m,3h),5.90(s,2h),5.14(s,2h).
13
c nmr(150mhz,dmso
‑
d6)δ165.83,153.02,147.30,140.94,129.09,128.90,126.85,116.84,114.29,113.52,108.08.esi
‑
ms:m/z 305.08(m+1).c
13
h
11
n3ose[304.22].
[0138]
实施例29:n
‑
(4
‑
(环丙烷磺酰胺)苯基)
‑2‑
碘
‑4‑
硝基苯甲酰胺(i
‑
21)的制备
[0139]
将中间体n
‑
(4
‑
氨基苯基)
‑2‑
碘
‑4‑
硝基苯甲酰胺(i
‑
18)(0.40g,1.04mmol,1.0eq.)溶于4ml的二氯甲烷中,加入三乙胺(0.18ml,1.25mmol,1.2eq.),冰浴条件下搅拌,随后将环丙烷磺酰氯(0.16g,1.14mmol,1.1eq.)溶于2.0ml的二氯甲烷中,缓慢加入上述溶液中,0℃条件下搅拌,tlc监测;6h后反应完毕,反应完毕,加入饱和碳酸氢钠溶液(3
×
6.0ml),二氯甲烷20ml萃取,取有机相;加入饱和氯化钠溶液(3
×
10ml)洗涤,有机相用无水硫酸钠干燥,过滤,滤液减压浓缩,所得粗品经硅胶柱层析分离(洗脱剂甲醇:二氯甲烷=1:100)得到n
‑
(4
‑
(环丙烷磺酰胺)苯基)
‑2‑
碘
‑4‑
硝基苯甲酰胺(i
‑
21)的粗品0.19g,淡黄色固体,产率38%。
[0140]
波谱数据:esi
‑
ms:m/z 486.04(m
‑
1).c
16
h
14
in3o5s[487.26].
[0141]
实施例30:n
‑
(4
‑
(6
‑
硝基
‑3‑
氧代苯并[d][1,2]偶氮硒
‑
2(3h)
‑
基)苯基)环丙烷磺酰胺(i
‑
22)的制备
[0142]
将中间体i
‑
21(0.15g,0.31mmol,1.0eq.)、硒氰酸钾(0.05g,0.37mmol,1.2eq.)、碘化亚铜(0.06g,0.31mmol,1.0eq.)、1,10
‑
菲啰啉(0.06g,0.31mmol,1.0eq.)、碳酸铯(0.25g,0.78mmol,2.5eq.)溶于6ml n,n
‑
二甲基甲酰胺,n2保护,100℃加热回流,tlc监测;反应完毕,加入6ml水,乙酸乙酯(3
×
10ml)萃取,取有机相,加入饱和氯化钠溶液(3
×
10ml)洗涤,有机相用无水硫酸钠干燥,过滤,滤液减压浓缩,所得粗品经硅胶柱层析(洗脱剂甲
醇:二氯甲烷=1:50)分离纯化后得到目标产物n
‑
(4
‑
(6
‑
硝基
‑3‑
氧代苯并[d][1,2]偶氮硒
‑
2(3h)
‑
基)苯基)环丙烷磺酰胺(i
‑
22)0.02g,棕色固体,产率14%,熔点:250
‑
252℃。
[0143]
波谱数据:1h nmr(400mhz,dmso
‑
d6)δ9.88(s,1h),8.94(d,j=1.8hz,1h),8.26(dd,j=8.5,1.9hz,1h),8.09(d,j=8.5hz,1h),7.62(d,j=8.7hz,2h),7.34(d,j=8.7hz,2h),2.68(p,j=6.5hz,1h),0.98(d,j=6.3hz,4h).
13
c nmr(151mhz,dmso
‑
d6)δ163.90,150.20,140.52,137.02,135.42,133.60,129.56,126.22,122.06,121.59,121.37,30.19,5.53.esi
‑
ms:m/z 461.89(m+na).c
13
h9n3o3se[438.32].
[0144]
实施例31:目标化合物的抗主蛋白酶(m
pro
)的实验
[0145]
实验原理:
[0146]
采用的荧光共振能量转移(fluorescence resonance energy transfer,fret)法,底物结构为:mca
‑
avlqsgfr
‑
lys(dnp)
‑
lys
‑
nh2,其中mca是荧光供体,dnp是荧光受体或称为荧光淬灭基团,完整的序列即含有荧光基团,又含有荧光淬灭基团,由于两个基团空间距离较近,淬灭基团的抑制作用使得荧光基团不会产生荧光。当加入sars
‑
cov
‑
2主蛋白酶m
pro
后,由于主蛋白酶能够在氨基酸q和s之间进行切割,使得荧光基团远离淬灭基团,在激发光为320nm,发射波长为405nm下产生荧光,通过测定荧光来检测m
pro
的活性,进而间接反应化合物的抑制活性(见附图1)(dai wenhao,et al.,science.368(6497):1331
‑
1335,2020.qiao jingxin,et al.,science.371(6536):1374
‑
1378,2021.)。
[0147]
实验方法:
[0148]
使用荧光共振能量转移法,测试了目标化合物的对主蛋白酶的抑制活性。使用mca
‑
avlqsgfr
‑
lys(dnp)
‑
lys
‑
nh2为反应底物。在避光条件下,将0.6μm的sars
‑
cov
‑
2 m
pro
,0.25μm的底物和1.0μm的化合物加入96孔板中进行初筛,37℃下反应10分钟,使用多功能酶标仪检测每组的荧光强度,激发波长为320nm,发射波长为405nm。实验分为空白对照组,阳性对照组和实验组。以依布硒啉作为实验阳性对照组,将化合物在1μm浓度下,抑制率>60%,淬灭率<20%的化合物进行复筛。实验初筛结果如下表所示。
[0149]
表2.目标化合物(二苯并异硒唑酮衍生物)抑制sars
‑
cov
‑
2主蛋白酶的初筛结果
[0150]
[0151][0152]
复筛:选取0.6μm的sars
‑
cov
‑
2 m
pro
,0.25μm的底物和四个浓度梯度(0.05
‑
1μm)测试化合物的ic
50
。另设表面活性剂(0.01%triton x
‑
100)排除化合物聚集影响mpro蛋白的活性。每组设置3个复孔,10℃下反应10分钟,使用多功能酶标仪检测每组的荧光强度,激发波长为320nm,发射波长405nm。实验结果如下表。
[0153]
表3.目标化合物(二苯并异硒唑酮衍生物)抑制sars
‑
cov
‑
2主蛋白酶的复筛结果
[0154]
[0155][0156]
a
ic
50
(μm):对酶的抑制达到50%时,所需化合物浓度,即半数抑制浓度;依布硒啉:已报道的一类sars
‑
cov
‑
2主蛋白酶抑制剂,作为阳性对照。
[0157]
实验结论分析:
[0158]
新合成的苯并异硒唑酮衍生物呈现出显著的抗sars
‑
cov
‑
2 m
pro
活性。经初步的活性筛选,4个化合物i
‑
13、i
‑
14、i
‑
15、i
‑
16的抗sars
‑
cov
‑
2 m
pro
抑制率>60%。进一步对这四个化合物进行复筛,结果显示化合物i
‑
14的活性与依布硒啉相当,化合物i
‑
13、i
‑
15、i
‑
16的活性优于先导化合物依布硒啉,具有进一步研究的价值。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1