异白叶藤碱类似物、从诺氟沙星到异白叶藤碱类似物的制备方法和应用
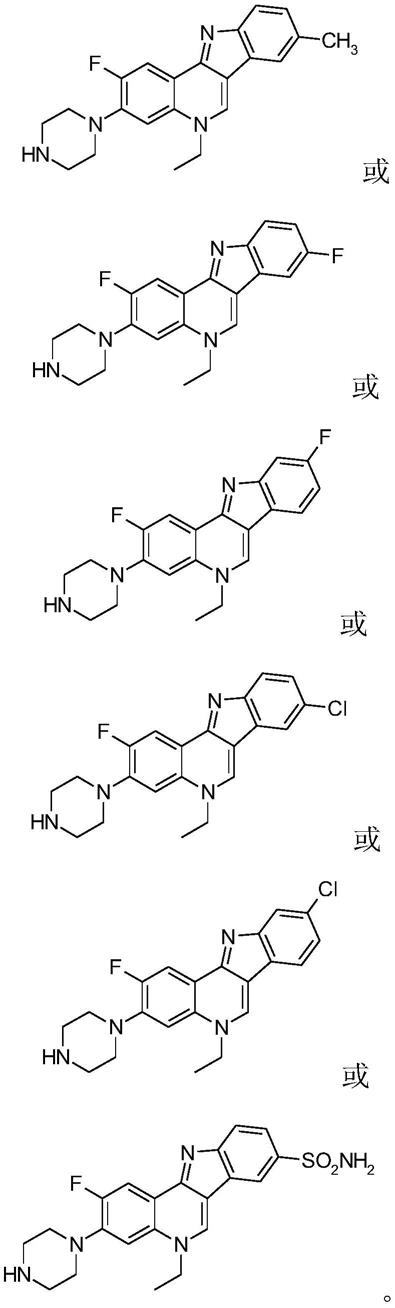
1.本发明涉及有机合成和新药物研发相关的药物化学的技术领域,尤其涉及一种异白叶藤碱类似物,同时还涉及一种从诺氟沙星到异白叶藤碱类似物的制备方法,以及其在制备抗结核药物中的应用。
背景技术:
2.结核病是由结核杆菌引起的高发病率的慢性传染疾病,由于缺乏有效的治疗药物,目前已成为全球所面临的一个紧迫的公共卫生和社会问题。同时,加之结核杆菌对现有药物易产生耐药性,尤其是多药耐药性的产生,对开发抗结核药物提出新挑战,导致自利福平抗结核药被发现以来的半个多世纪尚没有一个新型化合物用于结核病的治疗。因此,抗结核药物的研发是一项高耗时、高投入的复杂智力创新工程而被受关注。其中,以天然药物有效成分为先导物,对其结构进行优化修饰,是发现新药最经济有效的策略。在多种天然有效成分的研发中发现,以吲哚并喹啉为结构特征骨架的白叶藤碱类生物碱,如白叶藤碱(cryptolepine,a)、异白叶藤碱(isocryptolepine,b)、新白叶藤碱(neocryptolepine, c)等,
[0003][0004]
因结构独特,且具有较好的抗结疟原虫、抗肿瘤等多种生物活性而引起研究的兴趣,但是对其抗结核活性的报道尚少。然而,由于白叶藤类生物碱的来源困难,加之水溶性差而导致其生物利用度低等缺陷而限制临床应用。为此,如何以白叶藤类生物碱为先导物,以原子经济策略设计新型结构的吲哚并喹啉类抗结核药物显得极为重要。一方面,基于氟喹诺酮药物不仅是临床重要的抗菌药物,同时也是临床二线抗结核药物,如把白叶藤类生物碱及氟喹诺酮类的优势结构相拼合,发挥各自的药效团优势,有可能设计发现新型的吲哚并喹啉类抗结核药物;另一方面,通过引入氟喹诺酮药物分子中的有效取代基,进一步改善其药效学和药动学性质以克服现有生物碱的自身缺陷,促进新型吲哚并喹啉类抗结核药物的成药性发展。
技术实现要素:
[0005]
针对如何以白叶藤类生物碱为先导物,以原子经济策略设计新型结构的吲哚并喹啉类抗结核药物的技术问题,本发明提出一种异白叶藤碱类似物,同时还涉及一种从诺氟沙星到异白叶藤碱类似物的制备方法,以及其在制备抗结核药物中的应用。本发明从商业购得的氟喹诺酮药物诺氟沙星(ii)为原料,通过还原脱酸到喹啉酮(6
‑
氟
‑1‑
乙基
‑7‑
哌嗪
‑
1
‑
基
‑
2,3
‑
二氢
‑
喹啉
‑
4(1h)
‑
酮,iii),然后与苯肼类通过费歇尔吲哚合成方法成功构建了一种异白叶藤碱类似物;选择白叶藤碱类生物碱中的异白叶藤碱(b)为先导物,以其吲哚并[3,2
‑
c]喹啉为优势骨架,引入氟喹诺酮药物结构中的亲水性碱性哌嗪基以增加水溶性、改善其生物利用度,同时氟原子的引入可增加药物分子的渗透性以提高其生物活性。
[0006]
为了达到上述目的,本发明的技术方案是这样实现的:
[0007]
一种异白叶藤碱类似物,化学结构通式如式i所示:
[0008][0009]
式i中的取代基r可独立为氢原子
‑
h、甲氧基
‑
och3、甲基
‑
ch3、氟原子
‑
f、氯原子
‑
cl或黄酰胺基
‑
so2nh2。
[0010]
优选的,异白叶藤碱类似物的化学结构式为:
[0011]
[0012][0013]
一种从诺氟沙星到异白叶藤碱类似物的制备方法,包括以下步骤:
[0014]
s1、以式ii所示的诺氟沙星为原料与硼氢化钾通过还原脱羧反应制得如式 iii所示的2,3
‑
二氢喹啉
‑4‑
酮;
[0015]
[0016]
s2、以步骤s1中得到的2,3
‑
二氢喹啉
‑4‑
酮与苯肼类通过费歇尔吲哚合成法制得吲哚并喹啉母核结构特征的白叶藤碱类生物碱;
[0017][0018]
s3、将步骤s2得到的吲哚并喹啉母核结构特征的白叶藤碱类生物碱进行后处理得到如权利要求1或2的异白叶藤碱类似物。
[0019]
优选的,以商业得到的式ii所示的诺氟沙星为原料,经与商业购得的分析纯化学试剂硼氢化钠发生还原脱羧反应制得式iii所示6
‑
氟
‑1‑
乙基
‑7‑
哌嗪
‑1‑
基
ꢀ‑
2,3
‑
二氢
‑
喹啉
‑
4(1h)
‑
酮,iii,其制备方法参考文献(kondo h,sakamoto f,et al. studis on prodrugs.7.synthesis and antimicrobial activity of 3
‑
formylquinolonederivatives,j med chem,1988,31(1):221
‑
225.)的类似方法。
[0020][0021]
作为进一步的改进,用价格便宜、不易吸潮、反应温和的硼氢化钾替代文献中的硼氢化钠。
[0022]
步骤s1具体包括以下步骤:
[0023]
y1、取诺氟沙星与溶剂混合制成悬浮液,常温搅拌下向悬浮液内分次缓慢加入硼氢化钾,将混合反应物进行水浴加热,搅拌回流反应至诺氟沙星消失,获得混合液;
[0024]
y2、将步骤y1中获得的混合液放置至室温,使用蒸发器蒸除混合液中的溶剂,得到剩余物;
[0025]
y3、将步骤y2中的获得的剩余物加入去离子水中混合,使用浓盐酸调至ph≈2,加入活性炭脱色,使用质量浓度为30%的氢氧化钠调至ph≈10,放置析出固体;
[0026]
y4,将步骤y3中析出的固体依次用热水重结晶和正己烷重结晶得到2,3
‑ꢀ
二氢喹啉
‑4‑
酮。
[0027]
优选的,步骤s2具体包括以下步骤:
[0028]
t1、将步骤s1中得到的2,3
‑
二氢喹啉
‑4‑
酮溶解于无水乙醇中,加入苯肼类,常温搅拌至2,3
‑
二氢喹啉
‑4‑
酮消失,获得混合液;
[0029]
t2、向步骤t1中获得的混合液中滴加环合催化剂,进行加热回流反应,放置至室温,滤集产生的吲哚并喹啉母核结构特征的白叶藤碱类生物碱。
[0030]
优选的,步骤s3具体包括以下步骤:
[0031]
z1、将步骤s3中获得的吲哚并喹啉母核结构特征的白叶藤碱类生物碱溶于去离子水中,加入活性炭回流脱色,加入浓氨水调至ph≈10,滤集产生的固体;
[0032]
z2、将步骤t3中获得的固体用无水乙醇
‑
乙酸乙酯混合溶剂重结晶,得淡黄色结晶的异白叶藤碱类似物。
[0033]
优选的,步骤y1中的溶剂为无水甲醇、无水乙醇或95%的乙醇,诺氟沙星和硼氢化钾的摩尔比为1:(1~3)。
[0034]
优选的,步骤t1中的2,3
‑
二氢喹啉
‑4‑
酮和苯肼类的摩尔比为1:(1~2),苯肼类为苯肼、对甲基苯肼、间甲基苯肼、邻甲基苯肼、对氟苯肼、间氟苯肼或间氟磺酰胺基苯肼,步骤t2中的环合催化剂为浓盐酸、浓硫酸、磷酸、多聚磷酸、冰乙酸或三氟乙酸,回流反应的时间为10~24h。
[0035]
异白叶藤碱类似物在制备抗结核药物中的应用。
[0036]
优选的,异白叶藤碱类似物用于制备抑制结核分支杆菌药物,结核分支杆菌为h
37
ra或h
37
rv。
[0037]
本发明的有益效果:
[0038]
1.本发明的异白叶藤碱类似物,保留了异白叶藤碱的优势骨架—吲哚并喹啉母核,同时也兼有氟喹诺酮药物的特征结构—喹啉环,尤其是作为氟喹诺酮药物的有效修饰基,其亲水性的碱性哌嗪基作为吲哚并喹啉母核的修饰基,不仅可有效改善白叶藤生物碱类的水溶性,提高生物利用度,有利于成药性的发展,同时氟原子的引入可增加药物渗透作用,实现增效降毒及抗耐药的效果。
[0039]
2.本发明的异白叶藤碱类似物中具备的吲哚并喹啉母核和喹啉环,实现了不同结构药效团的互补和活性的叠加,实验例中的体外抗结核活性测试结果表明,所述化合物对测试结核菌株有较好的生长抑制活性,部分化合物的活性与对照异烟肼相当,且兼有抗耐药性和较低的细胞毒性,具有优秀的体外抑制结核杆菌生长活性,可作为新型的吲哚并喹啉结构特征的抗结核药物进一步开发。
[0040]
3.本发明的制备方法使用诺氟沙星通过还原脱羧反应与硼氢化钾制备2,3
‑ꢀ
二氢喹啉
‑4‑
酮,接着使用2,3
‑
二氢喹啉
‑4‑
酮与苯肼类通过费歇尔吲哚合成法制备异白叶藤碱类似物,实现了由氟喹诺酮结构到吲哚并喹啉骨架的有效化学构建,扩展了异白叶藤碱的结构修饰新途径,达到了氟喹诺酮药物与天然吲哚并喹啉类生物碱的优势结构的互补。
具体实施方式
[0041]
下面将结合本发明实施例,对本发明实施例中的技术方案进行清楚、完整地描述,显然,所描述的实施例仅仅是本发明一部分实施例,而不是全部的实施例。基于本发明中的实施例,本领域普通技术人员在没有付出创造性劳动前提下所获得的所有其他实施例,都属于本发明保护的范围。
[0042]
实施例1
[0043]
一种异白叶藤碱类似物,化学结构通式如式i所示:
1
‑
ch2),2.25~2.64(3h,m,3
‑
h和nh),1.22(3h,t,ch3);ms(m/z):278[m+h]
+
,计算(c
15
h
20
fn3o):277.34。
[0052]
s2、以步骤s1中得到的2,3
‑
二氢喹啉
‑4‑
酮与苯肼类通过费歇尔吲哚合成法制得吲哚并喹啉母核结构特征的白叶藤碱类生物碱。具体的说,包括以下步骤: t1、将步骤s1中得到的2,3
‑
二氢喹啉
‑4‑
酮溶解于溶剂中,加入苯肼类,常温搅拌至2,3
‑
二氢喹啉
‑4‑
酮消失,获得混合液;t2、向步骤t1中获得的混合液中滴加环合催化剂,进行加热回流反应,放置至室温,滤集产生的吲哚并喹啉母核结构特征的白叶藤碱类生物碱。
[0053]
在本实施例中,具体包括:取1
‑
乙基
‑6‑
氟
‑7‑
哌嗪
‑1‑
基
‑
2,3
‑
二氢
‑
喹啉
‑
4(1h)
‑ꢀ
酮1.0g(3.6mmol)溶解于15ml无水乙醇中,加入苯肼0.50g(4.6mmol),常温搅拌反应20h,有大量的沉淀物生成。加入浓盐酸(0.50ml),混合反应物回流反应12h,放置过夜。滤集产生的固体。固体即为吲哚并喹啉母核结构特征的白叶藤碱类生物碱。
[0054]
s3、将步骤s2得到的吲哚并喹啉母核结构特征的白叶藤碱类生物碱进行后处理得到如权利要求1或2所述的异白叶藤碱类似物。具体的说,包括以下步骤:z1、将步骤s3中获得的吲哚并喹啉母核结构特征的白叶藤碱类生物碱溶于去离子水中,加入活性炭回流脱色,加入浓氨水调至ph≈10,滤集产生的固体;
[0055]
z2、将步骤t3中获得的固体用无水乙醇
‑
乙酸乙酯混合溶剂重结晶,得淡黄色结晶的异白叶藤碱类似物。
[0056]
在本实施例中,具体包括:用50ml去离子水溶解固体,加入适量的活性炭,回流脱色1h。热过滤,滤液用氨水调ph≈10.0。滤集产生的固体,干燥,用无水乙醇
‑
乙酸乙酯混合溶剂(v:v=5:1)重结晶,得淡黄色结晶目标物式i
‑
1,产率52.4%,m.p.232~234℃。1h nmr(400mhz,cd3cl)δ:1.42(3h,t,ch3), 3.12~3.45(8h,m,哌嗪
‑
h),3.62(1h,br,nh),4.54(2h,q,ch2),7.17~8.12(6h, m,1
‑
h、4
‑
h和ph
‑
h),8.72(1h,s,6
‑
h);ms(m/z):349[m+h]
+
,计算(c
21
h
21
fn4): 348.43。
[0057]
实施例2
[0058]
一种异白叶藤碱类似物,本实施例与实施例1的区别在于式i中的取代基r 为甲氧基,则异白叶藤碱类似物即2
‑
氟
‑5‑
乙基
‑8‑
甲氧基
‑3‑
哌嗪
‑1‑
基
‑
5h
‑
吲哚并 [3,2
‑
c]喹啉的化学结构式为:
[0059][0060]
本实施例使用了一种从诺氟沙星到异白叶藤碱类似物的制备方法用于制备上述异白叶藤碱类似物(i
‑
2),具体包括以下步骤:取1
‑
乙基
‑6‑
氟
‑7‑
哌嗪
‑1‑
基
ꢀ‑
2,3
‑
二氢
‑
喹啉
‑
4(1h)
‑
酮1.0g(3.6mmol)溶解于15ml无水乙醇中,加入对甲氧基苯肼0.62g(4.5mmol),常温搅拌反应过夜,有明显的沉淀物生成。加入浓盐酸(0.50ml),混合反应物回流反应16h,放置过夜。滤集产生的固体,用50ml 去离子水溶解固体,加入适量的活性炭,回流脱色1h。热过滤,滤液用氨水调 ph≈10.0。滤集产生的固体,干燥,用无水乙醇
‑
乙酸乙酯混合溶剂
(v:v=5:1)重结晶,得淡黄色结晶目标物式i
‑
2,产率53.6%,m.p.233~235℃。1h nmr(400mhz, cd3cl)δ:1.45(3h,t,ch3),3.16~3.47(8h,m,哌嗪
‑
h),3.68(1h,br,nh), 3.86(3h,s,och3),4.62(2h,q,ch2),7.45~8.23(5h,m,1
‑
h、4
‑
h和ph
‑
h), 8.86(1h,s,6
‑
h);ms(m/z):376[m+h]
+
,计算(c
22
h
23
fn4o):378.45。
[0061]
值得说明的是,在本实施例中,制备1
‑
乙基
‑6‑
氟
‑7‑
哌嗪
‑1‑
基
‑
2,3
‑
二氢
‑
喹啉
ꢀ‑
4(1h)
‑
酮的方法与实施例1基本一致,区别在于本实施例中,将诺氟沙星和硼氢化钾的摩尔比设置为1:2,将实施例1中使用的无水甲醇作为溶剂替换为无水乙醇作为溶剂。
[0062]
实施例3
[0063]
一种异白叶藤碱类似物,本实施例与实施例1的区别在于式i中的取代基r 为甲氧基,则异白叶藤碱类似物即2
‑
氟
‑5‑
乙基
‑3‑
哌嗪
‑1‑
基
‑9‑
甲氧基
‑
5h
‑
吲哚并[3,2
‑
c]喹啉的化学结构式为:
[0064][0065]
本实施例使用了一种从诺氟沙星到异白叶藤碱类似物的制备方法用于制备上述异白叶藤碱类似物(i
‑
3),具体包括以下步骤:取1
‑
乙基
‑6‑
氟
‑7‑
哌嗪
‑1‑
基
ꢀ‑
2,3
‑
二氢
‑
喹啉
‑
4(1h)
‑
酮1.0g(3.6mmol)溶解于15ml无水乙醇中,加入间甲氧基苯肼0.83g(6.0mmol),常温搅拌反应24h,出现明显的沉淀物生成。加入浓盐酸(0.50ml),混合反应物回流反应12h,放置过夜。滤集产生的固体,用50ml 去离子水溶解固体,加入适量的活性炭,回流脱色1h。热过滤,滤液用氨水调 ph≈10.0。滤集产生的固体,干燥,用无水乙醇
‑
乙酸乙酯混合溶剂(v:v=6:1)重结晶,得淡黄色结晶目标物式i
‑
3,产率42.5%,m.p.234~236℃。1h nmr(400mhz, cd3cl)δ:1.44(3h,t,ch3),3.18~3.46(8h,m,哌嗪
‑
h),3.70(1h,br,nh), 3.88(3h,s,och3),4.64(2h,q,ch2),7.53~8.25(5h,m,1
‑
h、4
‑
h和ph
‑
h), 8.87(1h,s,6
‑
h);ms(m/z):376[m+h]
+
,计算(c
22
h
23
fn4o):378.45。
[0066]
值得说明的是,在本实施例中,制备1
‑
乙基
‑6‑
氟
‑7‑
哌嗪
‑1‑
基
‑
2,3
‑
二氢
‑
喹啉
ꢀ‑
4(1h)
‑
酮的方法与实施例1基本一致,区别在于本实施例中,将诺氟沙星和硼氢化钾的摩尔比设置为1:1,将实施例1中使用的无水甲醇作为溶剂替换为95%的乙醇作为溶剂。
[0067]
实施例4
[0068]
一种异白叶藤碱类似物,本实施例与实施例1的区别在于式i中的取代基r 为甲氧基,则异白叶藤碱类似物即2
‑
氟
‑5‑
乙基
‑3‑
哌嗪
‑1‑
基
‑
10
‑
甲氧基
‑
5h
‑
吲哚并[3,2
‑
c]喹啉的化学结构式为:
[0069][0070]
本实施例使用了一种从诺氟沙星到异白叶藤碱类似物的制备方法用于制备上述异白叶藤碱类似物(i
‑
4),具体包括以下步骤:取1
‑
乙基
‑6‑
氟
‑7‑
哌嗪
‑1‑
基
ꢀ‑
2,3
‑
二氢
‑
喹啉
‑
4(1h)
‑
酮1.0g(3.6mmol)溶解于15ml无水乙醇中,加入邻甲氧基苯肼0.72g(5.0mmol),常温搅拌反应24h,有沉淀物生成。加入浓盐酸(0.50 ml),混合反应物回流反应12h,放置过夜。滤集产生的固体,用50ml去离子水溶解固体,加入适量的活性炭,回流脱色1h。热过滤,滤液用氨水调ph≈10.0。滤集产生的固体,干燥,用无水乙醇
‑
乙酸乙酯混合溶剂(v:v=3:1)重结晶,得淡黄色结晶目标物式i
‑
4,产率40.2%,m.p.226~228℃。1h nmr(400mhz,cd3cl) δ:1.45(3h,t,ch3),3.23~3.47(8h,m,哌嗪
‑
h),3.65(1h,br,nh),3.86(3h, s,och3),4.62(2h,q,ch2),7.55~8.26(5h,m,1
‑
h、4
‑
h和ph
‑
h),8.86(1h, s,6
‑
h);ms(m/z):376[m+h]
+
,计算(c
22
h
23
fn4o):378.45。
[0071]
值得说明的是,在本实施例中,制备1
‑
乙基
‑6‑
氟
‑7‑
哌嗪
‑1‑
基
‑
2,3
‑
二氢
‑
喹啉
ꢀ‑
4(1h)
‑
酮的方法与实施例1基本一致,区别在于本实施例中,将诺氟沙星和硼氢化钾的摩尔比设置为1:2,将实施例1中使用的无水甲醇作为溶剂替换为95%的乙醇作为溶剂。
[0072]
实施例5
[0073]
一种异白叶藤碱类似物,本实施例与实施例1的区别在于式i中的取代基r 为甲基,则异白叶藤碱类似物即2
‑
氟
‑3‑
哌嗪
‑1‑
基
‑5‑
乙基
‑8‑
甲基
‑
5h
‑
吲哚并[3, 2
‑
c]喹啉的化学结构式为:
[0074][0075]
本实施例使用了一种从诺氟沙星到异白叶藤碱类似物的制备方法用于制备上述异白叶藤碱类似物(i
‑
5),具体包括以下步骤:取式ⅲ所示的1
‑
乙基
‑6‑
氟
‑7‑ꢀ
哌嗪
‑1‑
基
‑
2,3
‑
二氢
‑
喹啉
‑
4(1h)
‑
酮1.0g(3.6mmol)溶解于15ml无水乙醇中,加入对甲基苯肼0.70g(5.7mmol),常温搅拌反应过夜,有沉淀物生成。加入浓盐酸(0.50ml),混合反应物回流反应24h,放置过夜。滤集产生的固体,用50ml 去离子水溶解固体,加入适量的活性炭,回流脱色1h。热过滤,滤液用氨水调 ph≈10.0。滤集产生的固体,干燥,用无水乙醇
‑
乙酸乙酯混合溶剂(v:v=3:1)重结晶,得淡黄色结晶目标物式i
‑
5,产率36.4%,m.p.232~234℃。1h nmr(400mhz, cd3cl)δ:1.41(3h,t,ch3),2.27(3h,s,ph
‑
ch3),3.05~3.37(8h,m,哌嗪
‑
h), 3.62(1h,br,nh),4.57(2h,q,ch2),7.42~8.18(5h,m,1
‑
h、4
‑
h和ph
‑
h), 8.86(1h,s,6
‑
h);
ms(m/z):363[m+h]
+
,计算(c
22
h
23
fn4):362.45。
[0076]
值得说明的是,在本实施例中,制备1
‑
乙基
‑6‑
氟
‑7‑
哌嗪
‑1‑
基
‑
2,3
‑
二氢
‑
喹啉
ꢀ‑
4(1h)
‑
酮的方法与实施例1基本一致,区别在于本实施例中,将诺氟沙星和硼氢化钾的摩尔比设置为1:3,将实施例1中使用的无水甲醇作为溶剂替换为无水乙醇作为溶剂。
[0077]
实施例6
[0078]
一种异白叶藤碱类似物,本实施例与实施例1的区别在于式i中的取代基r 为f原子,则异白叶藤碱类似物即2,8
‑
二氟
‑3‑
哌嗪
‑1‑
基
‑5‑
乙基
‑
5h
‑
吲哚并[3, 2
‑
c]喹啉的化学结构式为:
[0079][0080]
本实施例使用了一种从诺氟沙星到异白叶藤碱类似物的制备方法用于制备上述异白叶藤碱类似物(i
‑
6),具体包括以下步骤:取1
‑
乙基
‑6‑
氟
‑7‑
哌嗪
‑1‑
基
ꢀ‑
2,3
‑
二氢
‑
喹啉
‑
4(1h)
‑
酮1.0g(3.6mmol)溶解于15ml无水乙醇中,加入对氟苯肼0.60g(4.8mmol),常温搅拌反应24h,有大量的沉淀物生成。加入浓盐酸(0.50 ml),混合反应物回流反应20h,放置过夜。滤集产生的固体,用50ml去离子水溶解固体,加入适量的活性炭,回流脱色1h。热过滤,滤液用氨水调ph≈10.0。滤集产生的固体,干燥,用无水乙醇
‑
乙酸乙酯混合溶剂(v:v=6:1)重结晶,得淡黄色结晶目标物式i
‑
6,产率52.4%,m.p.241~243℃。1h nmr(400mhz,cd3cl) δ:1.52(3h,t,ch3),3.31~3.52(8h,m,哌嗪
‑
h),3.72(1h,br,nh),4.70(2h, q,ch2),7.62~8.26(5h,m,1
‑
h、4
‑
h和ph
‑
h),8.90(1h,s,6
‑
h);ms(m/z): 367[m+h]
+
,计算(c
21
h
20
f2n4):366.42。
[0081]
实施例7
[0082]
一种异白叶藤碱类似物,本实施例与实施例1的区别在于式i中的取代基r 为f原子,则异白叶藤碱类似物即2,9
‑
二氟
‑3‑‑
哌嗪
‑1‑
基
‑5‑
乙基
‑
5h
‑
吲哚并[3, 2
‑
c]喹啉的化学结构式为:
[0083][0084]
本实施例使用了一种从诺氟沙星到异白叶藤碱类似物的制备方法用于制备上述异白叶藤碱类似物(i
‑
7),具体包括以下步骤:取1
‑
乙基
‑6‑
氟
‑7‑
哌嗪
‑1‑
基
ꢀ‑
2,3
‑
二氢
‑
喹啉
‑
4(1h)
‑
酮1.0g(3.6mmol)溶解于15ml无水乙醇中,加入间氟苯肼0.66g(5.2mmol),常温搅拌反应过夜,有明显的沉淀物生成。加入浓盐酸(0.50 ml),混合反应物回流反应16h,放置过夜。滤集产生的固体,用50ml去离子水溶解固体,加入适量的活性炭,回流脱色1h。热过
滤,滤液用氨水调ph≈10.0。滤集产生的固体,干燥,用无水乙醇
‑
乙酸乙酯混合溶剂(v:v=5:1)重结晶,得淡黄色结晶目标物式i
‑
7,产率42.6%,m.p.230~232℃。1h nmr(400mhz,cd3cl) δ:1.48(3h,t,ch3),3.33~3.62(8h,m,哌嗪
‑
h),3.73(1h,br,nh),4.72(2h, q,ch2),7.58~8.25(5h,m,1
‑
h、4
‑
h和ph
‑
h),8.91(1h,s,6
‑
h);ms(m/z): 367[m+h]
+
,计算(c
21
h
20
f2n4):366.42。
[0085]
实施例8
[0086]
一种异白叶藤碱类似物,本实施例与实施例1的区别在于式i中的取代基r 为氯原子,则异白叶藤碱类似物即2
‑
氟
‑8‑
氯
‑3‑
哌嗪
‑1‑
基
‑5‑
乙基
‑
5h
‑
吲哚并[3, 2
‑
c]喹啉的化学结构式为:
[0087][0088]
本实施例使用了一种从诺氟沙星到异白叶藤碱类似物的制备方法用于制备上述异白叶藤碱类似物(i
‑
8),具体包括以下步骤:取1
‑
乙基
‑6‑
氟
‑7‑
哌嗪
‑1‑
基
ꢀ‑
2,3
‑
二氢
‑
喹啉
‑
4(1h)
‑
酮1.0g(3.6mmol)溶解于15ml无水乙醇中,加入对氯苯肼0.74g(5.2mmol),常温搅拌反应24h,有大量的沉淀物生成。加入浓盐酸(0.50 ml),混合反应物回流反应24h,放置过夜。滤集产生的固体,用50ml去离子水溶解固体,加入适量的活性炭,回流脱色1h。热过滤,滤液用氨水调ph≈10.0。滤集产生的固体,干燥,用无水乙醇
‑
乙酸乙酯混合溶剂(v:v=5:1)重结晶,得淡黄色结晶目标物式i
‑
8,产率41.5%,m.p.232~234℃。1h nmr(400mhz,cd3cl) δ:1.43(3h,t,ch3),3.27~3.60(8h,m,哌嗪
‑
h),3.68(1h,br,nh),4.70(2h, q,ch2),7.47~8.24(5h,m,1
‑
h、4
‑
h和ph
‑
h),8.89(1h,s,6
‑
h);ms(m/z): 383(cl
35
)[m+h]
+
,385(cl
37
)[m+h]
+
,计算(c
21
h
20
clfn4):382.87。
[0089]
实施例9
[0090]
一种异白叶藤碱类似物,本实施例与实施例1的区别在于式i中的取代基r 为氯原子,则异白叶藤碱类似物即2
‑
氟
‑9‑
氯
‑3‑
哌嗪
‑1‑
基
‑5‑
乙基
‑
5h
‑
吲哚并[3, 2
‑
c]喹啉的化学结构式为:
[0091][0092]
本实施例使用了一种从诺氟沙星到异白叶藤碱类似物的制备方法用于制备上述异白叶藤碱类似物(i
‑
9),具体包括以下步骤:取1
‑
乙基
‑6‑
氟
‑7‑
哌嗪
‑1‑
基
ꢀ‑
2,3
‑
二氢
‑
喹啉
‑
4(1h)
‑
酮1.0g(3.6mmol)溶解于15ml无水乙醇中,加入间氯苯肼0.70g(5.0mmol),常温搅拌反应24h,有明显的沉淀物生成。加入浓盐酸(0.50 ml),混合反应物回流反应16h,放置
过夜。滤集产生的固体,用50ml去离子水溶解固体,加入适量的活性炭,回流脱色1h。热过滤,滤液用氨水调ph≈10.0。滤集产生的固体,干燥,用无水乙醇
‑
乙酸乙酯混合溶剂(v:v=6:1)重结晶,得淡黄色结晶目标物式i
‑
9,产率41.3%,m.p.231~233℃。1h nmr(400mhz,cd3cl) δ:1.48(3h,t,ch3),3.30~3.67(8h,m,哌嗪
‑
h),3.72(1h,br,nh),4.73(2h, q,ch2),7.50~8.26(5h,m,1
‑
h、4
‑
h和ph
‑
h),8.90(1h,s,6
‑
h);ms(m/z): 383(cl
35
)[m+h]
+
,385(cl
37
)[m+h]
+
,计算(c
21
h
20
clfn4):382.87。
[0093]
实施例10
[0094]
一种异白叶藤碱类似物,本实施例与实施例1的区别在于式i中的取代基r 为磺酰胺基,则异白叶藤碱类似物即2
‑
氟
‑8‑
磺酰胺基
‑3‑
哌嗪
‑1‑
基
‑5‑
乙基
‑
5h
‑ꢀ
吲哚并[3,2
‑
c]喹啉的化学结构式为:
[0095][0096]
本实施例使用了一种从诺氟沙星到异白叶藤碱类似物的制备方法用于制备上述异白叶藤碱类似物(i
‑
10),具体包括以下步骤:取1
‑
乙基
‑6‑
氟
‑7‑
哌嗪
‑1‑ꢀ
基
‑
2,3
‑
二氢
‑
喹啉
‑
4(1h)
‑
酮1.0g(3.6mmol)溶解于15ml无水乙醇中,加入对间氯磺酰胺基苯肼0.67g(3.6mmol),常温搅拌反应过夜,有大量的沉淀物生成。加入浓盐酸(0.50ml),混合反应物回流反应24h,放置过夜。滤集产生的固体,用50ml去离子水溶解固体,加入适量的活性炭,回流脱色1h。热过滤,滤液用氨水调ph≈10.0。滤集产生的固体,干燥,用无水乙醇
‑
乙酸乙酯混合溶剂 (v:v=5:1)重结晶,得淡黄色结晶目标物式i
‑
10,产率53.6%,m.p.244~246℃。1h nmr(400mhz,cd3cl)δ::1.57(3h,t,ch3),3.37~3.78(8h,m,哌嗪
‑
h), 3.75(1h,br,nh),4.82(2h,q,ch2),7.32~8.56(7h,m,1
‑
h、4
‑
h、ph
‑
h和 nh2),9.08(1h,s,6
‑
h);ms(m/z):428[m+h]
+
,计算(c
21
h
22
fn5o2s):427.50。
[0097]
值得说明的是,本发明还提供了异白叶藤碱类似物在制备抗结核药物中的应用,具体指异白叶藤碱类似物用于制备抑制结核分支杆菌药物,结核分支杆菌为h
37
ra或h
37
rv,下述对异白叶藤碱类似物在制备抗结核药物中的应用的实验例进行详细描述。
[0098]
实验例
[0099]
一、实施例1~10提供的异白叶藤碱类似物的体外抗结核菌活性测定
[0100]
1、实验试剂
[0101]
阳性对照品异烟肼(isoniazide,inh)和诺氟沙星(norfloxacin)购自河南省食品药品检验所;7h9液体培养液购自美国difco公司。在无菌条件下,用二甲基亚砜(dmso)将阳性对照品及实施例i
‑
1~i
‑
10供试品配成4mg/ml的溶液,超声充分溶解后以0.22μm滤膜过滤,滤液作为储备液置
‑
20℃下保存待用(使用时,为避免dmso对实验结果的影响,dmso在培养液中的浓度<0.5%)。
[0102]
2、结核菌株
[0103]
实验用结核菌株分别为结核分枝杆菌标准株h
37
ra(atcc25177)、 h
37
rv
(atcc27294)及3株临床分离的耐药结核分枝杆菌编号分别为h6、h7 和h10,均由河南省疾病预防控制中心提供并提供实验数据的测定。其中,h6、 h7为对异烟肼、利福平、乙胺丁醇、链霉素、氧氟沙星的多重耐药菌株,h10 为对异烟肼、利福平的耐药菌株。
[0104]
3、实验方法
[0105]
1)菌株悬液的制备:将培养2~3周菌龄的待测结核杆菌用接种环取出接入灭菌小瓶中,混均至成乳状,生理盐水稀释,通过与no.1麦氏标准比浊管比浊,将菌液配成1mg/ml的菌液,再用生理盐水稀释至1
×
105cfu备用。
[0106]
2)在96孔培养板上,加入200μml合适浓度的待测化合物溶液(以无菌的 7h9液体培养液稀释待测化合物至200μg/ml),然后根据需要再对待测化合物进行稀释(倍比稀释至50、25、12.5、6.25、3.125、1.56、0.78、0.39、0.195、0.097、 0.048、0.024、0.012μg/ml),并设无药对照孔。
[0107]
3)将上述稀释好的菌液加入到所有检测孔中以及无药对着孔中,将这些板至于恒温孵箱中,每板在37℃,5%co2条件下培养21天。40
×
的显微镜观察,肉眼未见菌株生长的最低浓度即为该药的最低抑菌浓度(mic)。同时,以异烟肼、诺氟沙星为阳性对照,以dmso和不添加任何化合物的培养液作为阴性对照。每个数据平行测定三次,求其平均值,实验结果见表1所示。
[0108]
表1供试样品的体外抗结核活性(mic)
[0109][0110]
表1结果表明,实施例1~10提供的化合物中,实施例1、实施例4、实施例6、实施例7和实施例10对h
37
ra和h
37
rv两种结核分枝杆菌标准株的mic值低于对照诺氟沙星,尤其是实施例7和实施例10的活性与异烟肼相当,表现出较好的体外抗结核菌活性。同时,实施例1~10提供的化合物对3种临床分离的耐药菌株h6、h7和h10的mic值远低于对照诺氟沙星或异烟肼,体现出较好的抗耐药活性。
[0111]
二、实施例1~10的体外细胞毒性测定
[0112]
1、实验试剂
[0113]
阳性对照品异烟肼(isoniazide,inh)和诺氟沙星购自河南省食品药品检验所;正常细胞为非洲绿猴肾细胞株vero,购买于上海通派生物科技有限公司。 rpmi,胰蛋白酶
(trgpsin)和胎牛血清购于杭州四季青生物工程材料有限公司;溴化
‑
(4,5)
‑
二甲基
‑2‑
噻唑
‑
2,5
‑
二苯基四氮唑(mtt,amresco分装);十二烷基磺酸钠(sds),磷酸二氢钠购于天津科密欧化学试剂开发中心;乙二胺四乙酸二钠盐(edta)和二甲基亚砜(dmso)购于天津德恩化学式有限公司。
[0114]
2、实验供试液的配制
[0115]
在无菌条件下,用二甲基亚砜(dmso)将阳性对照品及实施例i
‑
1~i
‑
10供试品配制成1.0
×
10
‑4mol
·
l
‑1浓度的12种储备液,之后用质量百分比浓度为10%的小牛血清的rpmi
‑
1640培养液将储备液稀释成具有5个浓度梯度(0.1、1.0、5.0、 10.0、50.0μmol
·
l
‑1)的工作液,超声充分溶解后以0.22μm滤膜过滤,滤液作为供试液置
‑
20℃下保存待用。
[0116]
3、实验方法(mtt法)
[0117]
取对数生长期的vero非洲绿猴肾细胞株,以每孔6000个细胞接种于96 孔板,随后分别加入上述12种样品的具有5个浓度梯度的工作液,48小时后每孔加入5g
·
l
–1mtt(噻唑蓝)溶液10μl,继续再培养4小时之后加入100μl 质量百分比浓度为10%的十二烷基硫酸钠(sds)溶液。培养24小时,然后用酶标仪在570nm波长处测定吸光度(od)值。细胞增殖抑制率按公式计算:
[0118]
抑制率=[(1-实验组od值)/对照组od值]
×
100%
[0119]
然后以各供试样品的各浓度的对数值对各浓度对应的vero细胞抑制率作线性回归,得到剂量
‑
效应方程,从所得剂量
‑
效应方程计算出供试样品对实验vero 细胞的半数抑制浓度(ic
50
);每个数据平行测定三次,求其平均值,结果见表2 所示。
[0120]
表2供试样品的体外vero细胞毒性测定(ic
50
)
[0121][0122]
表2结果表明,实施例1~10提供的化合物对vero细胞的半数生长抑制浓度(ic
50
)阳性对照诺氟沙星相当,体现出较低的细胞毒作用,同时,实施例1~10 提供的化合物的ic
50
高于阳性异烟肼的ic
50
值,表明实施例1~10提供的化合物其细胞毒性低于阳性对照异烟肼。
[0123]
综上所述,实施例1~10提供的化合物体外不仅具有较好的抗结核菌活性,同时也具有潜在的抗耐药活性,且表现出较低的细胞毒作用。基于新药研究的规律,本发明的一种从诺氟沙星到异白叶藤碱类似物有望开发出高效低毒的抗结核药物。
[0124]
以上所述仅为本发明的较佳实施例而已,并不用以限制本发明,凡在本发明的精神和原则之内,所作的任何修改、等同替换、改进等,均应包含在本发明的保护范围之内。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1